Soil erodibility,microbial biomass,and physical-chemical property changes during long-term natural vegetation restoration:a case study in the Loess Plateau, China
1
2010
... 荒漠地区气候条件极端恶劣,雨水稀少、土壤有机质和养分贫瘠、土地退化等都对区域生态系统稳定和生物多样性构成严重威胁[1].通过生物措施改良改善土壤质地,提升土地肥力,进行生态植被恢复,是防治荒漠化、退化土地恢复最有效和可持续的手段[2].广泛分布于中国荒漠沙地的锦鸡儿属(Caragana)植物根系萌蘖能力强、主根深入地下、抗逆性强,并有与土壤中的固氮菌结合共生固氮效能高的特点,在改善土壤肥力、固持土壤、防风固沙、植被恢复中有重要的作用,能够作为荒漠沙地生态恢复的先锋物种[3]. ...
Changes in soil nutrient and enzyme activities under different vegetations in the Loess Plateau area,northwest China
1
2012
... 荒漠地区气候条件极端恶劣,雨水稀少、土壤有机质和养分贫瘠、土地退化等都对区域生态系统稳定和生物多样性构成严重威胁[1].通过生物措施改良改善土壤质地,提升土地肥力,进行生态植被恢复,是防治荒漠化、退化土地恢复最有效和可持续的手段[2].广泛分布于中国荒漠沙地的锦鸡儿属(Caragana)植物根系萌蘖能力强、主根深入地下、抗逆性强,并有与土壤中的固氮菌结合共生固氮效能高的特点,在改善土壤肥力、固持土壤、防风固沙、植被恢复中有重要的作用,能够作为荒漠沙地生态恢复的先锋物种[3]. ...
2
2011
... 荒漠地区气候条件极端恶劣,雨水稀少、土壤有机质和养分贫瘠、土地退化等都对区域生态系统稳定和生物多样性构成严重威胁[1].通过生物措施改良改善土壤质地,提升土地肥力,进行生态植被恢复,是防治荒漠化、退化土地恢复最有效和可持续的手段[2].广泛分布于中国荒漠沙地的锦鸡儿属(Caragana)植物根系萌蘖能力强、主根深入地下、抗逆性强,并有与土壤中的固氮菌结合共生固氮效能高的特点,在改善土壤肥力、固持土壤、防风固沙、植被恢复中有重要的作用,能够作为荒漠沙地生态恢复的先锋物种[3]. ...
... 锦鸡儿属植物隶属于豆科、蝶形花亚科、山羊豆族、黄芪亚族,多为落叶灌木、半灌木,稀为小乔木.本属物种演化随着气候和地理类型的变化,具有一定的规律性.周道玮等[4]依据形态特征的差异性,认为该属植物总体沿着寒冷和干旱两个方向分布定植:一支演化为所有叶轴宿存、子房被毛的寒化特征;另一支则演化出所有叶片假掌状的旱化特征.这种植株形态性状特征的演化主要是气候和地理环境的胁迫下分化、适应的结果.本研究选取的锦鸡儿属为亚湿润半干旱地区的优势豆科植物.该属植物根系大多能够与土壤中广泛存在的根瘤菌结合形成具有固氮能力的根瘤,将土壤空气中的氮素固定为植物可以吸收利用的氨[3]. ...
豆科锦鸡儿属(Caragana Fabr.)植物地理分布与分化研究
1
2005
... 锦鸡儿属植物隶属于豆科、蝶形花亚科、山羊豆族、黄芪亚族,多为落叶灌木、半灌木,稀为小乔木.本属物种演化随着气候和地理类型的变化,具有一定的规律性.周道玮等[4]依据形态特征的差异性,认为该属植物总体沿着寒冷和干旱两个方向分布定植:一支演化为所有叶轴宿存、子房被毛的寒化特征;另一支则演化出所有叶片假掌状的旱化特征.这种植株形态性状特征的演化主要是气候和地理环境的胁迫下分化、适应的结果.本研究选取的锦鸡儿属为亚湿润半干旱地区的优势豆科植物.该属植物根系大多能够与土壤中广泛存在的根瘤菌结合形成具有固氮能力的根瘤,将土壤空气中的氮素固定为植物可以吸收利用的氨[3]. ...
Fine-root distribution,production,decomposition,and effect on soil organic carbon of three revegetation shrub species in northwest China
3
2016
... 在生态环境整体恶劣的荒漠地区,植物群落的定植生长能够起到防风固沙的功效,尤以植物发达延伸生长的根系能够固持和改良土壤[5].土壤作为植物生长的载体,不仅为植株生长提供养分支持,同时也在植物固定外来碳源的作用下,改良提升自身的理化和生物特性[6],而这其中土壤微生物发挥着重要的作用.荒漠植物群落的适应性很大程度上受土壤微生物的影响[7-8].植物的生长与土壤微生物密切相关,土壤微生物群落代表了迄今为止世界上已知最大的生物多样性库[9].根际是受根系分泌物影响的狭窄土壤微生态位,每克根可含有多达1011个微生物菌体[10]及3万多种原核生物[11].越来越多的证据表明,这种根际微生物群落在决定相关植物健康和功能等方面具有重要作用,涉及植物根系内生菌群落及其遗传因素和多种环境因子综合的交互性作用[9-14].如根际土壤微生物群落对植物抗病害方面的影响最为显著,大多数在土壤中传播的病原体需要在根际腐生生长才能够接触到宿主或在宿主周围达到足够数量,进而有效地感染宿主并脱离根际微域,而病原体能否成功感染宿主植物,受根际土壤微生物群落的显著影响[15].还有研究指出,根际微生物群落对宿主植物的免疫系统具有调节作用[16].而植物内生微生物是指寄生在植物组织内至少一段时间的细菌、古生菌、真菌或病毒群落,对寄主无任何不利影响[17].它们在促进植物的生长发育、营养储备和抵御生物或非生物胁迫(如疾病、虫害、高温、盐或干旱)等方面起着关键作用[18-20].豆科植物与根瘤菌的共生关系通过生物固氮和协同进化对于改善土壤质地和肥力具有巨大潜力[21].Bulgarelli等[22]发现,一些植物内生菌是从根际土向植物根系内迁移而来的,并主要受到根际土壤因子的影响. ...
... 根际土中微生物群落的基因组比植物丰富,也被称为植物的第二基因组[9].根际微生物在调节植物个体的健康、适应性和群落生产力等方面都发挥着难以替代的作用[9,23-26],而植物群落又通过影响土壤养分循环等生态过程影响土壤微生物群落[6].此外,部分根际微生物可以进入植物根系组织形成植物内生菌群落,其群落结构组成与根际土壤微生物群落又有显著的差异性[27-28],这种细菌群落多样性和结构组成在根系组织内和根际土之间的显著差异现象被定义为“根际效应”[29].Sun等[30]发现,毛乌素沙地旱生灌丛下和灌丛间的土壤细菌群落也出现在了细菌群落结构组成显著差异的现象.锦鸡儿属植物对荒漠环境的适应能力和生态恢复作用始终都受到高度的关注[5,31-32].基于以上的研究结论,本研究的主旨设定为:①荒漠豆科锦鸡儿属灌丛下的4个根系微域(根系、根际土、根区土、灌丛间空白土)存在细菌群落多样性和结构组成的层级差异性变化;②探究荒漠豆科锦鸡儿属植物灌丛下根系微域细菌群落结构的层级变异性,及其发生变异的成因. ...
... 该研究于2018年9月在位于毛乌素沙地南缘的宁夏盐池毛乌素沙地生态系统国家定位观测研究站(37°04′—38°10′N,106°30′—107°47′E)开展.研究站位于半旱区,属典型的大陆性季风气候,常年干燥温暖.年平均气温8.1 ℃,日间气温和相对湿度分别是-29.4~37.5 ℃和49%~55%.多年平均降雨量约为292 mm.降雨集中在7—9月,占年降雨量的60%~70%.当地的土壤类型主要为风成沙质土[31].研究区0~1 m剖面土壤质地为砂质,平均容重1.5 g·cm-3.这一地区的生态退化主要是过度放牧、气候变化和风蚀造成的,干旱草原退化为目前的沙地.现在的植被恢复是在2001年通过飞播(油蒿、蒙古岩黄耆和细枝岩黄耆)、植苗扦插种植(小叶锦鸡儿和柠条锦鸡儿)和自然封育建立的[5].目前该区的主要优势旱生灌木种包括油蒿、柠条锦鸡儿、小叶锦鸡儿、蒙古岩黄耆、细枝岩黄耆、沙柳等. ...
Influence of plant species on physical,chemical and biological soil properties in a Mediterranean forest soil
2
2010
... 在生态环境整体恶劣的荒漠地区,植物群落的定植生长能够起到防风固沙的功效,尤以植物发达延伸生长的根系能够固持和改良土壤[5].土壤作为植物生长的载体,不仅为植株生长提供养分支持,同时也在植物固定外来碳源的作用下,改良提升自身的理化和生物特性[6],而这其中土壤微生物发挥着重要的作用.荒漠植物群落的适应性很大程度上受土壤微生物的影响[7-8].植物的生长与土壤微生物密切相关,土壤微生物群落代表了迄今为止世界上已知最大的生物多样性库[9].根际是受根系分泌物影响的狭窄土壤微生态位,每克根可含有多达1011个微生物菌体[10]及3万多种原核生物[11].越来越多的证据表明,这种根际微生物群落在决定相关植物健康和功能等方面具有重要作用,涉及植物根系内生菌群落及其遗传因素和多种环境因子综合的交互性作用[9-14].如根际土壤微生物群落对植物抗病害方面的影响最为显著,大多数在土壤中传播的病原体需要在根际腐生生长才能够接触到宿主或在宿主周围达到足够数量,进而有效地感染宿主并脱离根际微域,而病原体能否成功感染宿主植物,受根际土壤微生物群落的显著影响[15].还有研究指出,根际微生物群落对宿主植物的免疫系统具有调节作用[16].而植物内生微生物是指寄生在植物组织内至少一段时间的细菌、古生菌、真菌或病毒群落,对寄主无任何不利影响[17].它们在促进植物的生长发育、营养储备和抵御生物或非生物胁迫(如疾病、虫害、高温、盐或干旱)等方面起着关键作用[18-20].豆科植物与根瘤菌的共生关系通过生物固氮和协同进化对于改善土壤质地和肥力具有巨大潜力[21].Bulgarelli等[22]发现,一些植物内生菌是从根际土向植物根系内迁移而来的,并主要受到根际土壤因子的影响. ...
... 根际土中微生物群落的基因组比植物丰富,也被称为植物的第二基因组[9].根际微生物在调节植物个体的健康、适应性和群落生产力等方面都发挥着难以替代的作用[9,23-26],而植物群落又通过影响土壤养分循环等生态过程影响土壤微生物群落[6].此外,部分根际微生物可以进入植物根系组织形成植物内生菌群落,其群落结构组成与根际土壤微生物群落又有显著的差异性[27-28],这种细菌群落多样性和结构组成在根系组织内和根际土之间的显著差异现象被定义为“根际效应”[29].Sun等[30]发现,毛乌素沙地旱生灌丛下和灌丛间的土壤细菌群落也出现在了细菌群落结构组成显著差异的现象.锦鸡儿属植物对荒漠环境的适应能力和生态恢复作用始终都受到高度的关注[5,31-32].基于以上的研究结论,本研究的主旨设定为:①荒漠豆科锦鸡儿属灌丛下的4个根系微域(根系、根际土、根区土、灌丛间空白土)存在细菌群落多样性和结构组成的层级差异性变化;②探究荒漠豆科锦鸡儿属植物灌丛下根系微域细菌群落结构的层级变异性,及其发生变异的成因. ...
Soil Microbial diversity in the vicinity of a Negev Desert shrub-Reaumuria negevensis
1
2011
... 在生态环境整体恶劣的荒漠地区,植物群落的定植生长能够起到防风固沙的功效,尤以植物发达延伸生长的根系能够固持和改良土壤[5].土壤作为植物生长的载体,不仅为植株生长提供养分支持,同时也在植物固定外来碳源的作用下,改良提升自身的理化和生物特性[6],而这其中土壤微生物发挥着重要的作用.荒漠植物群落的适应性很大程度上受土壤微生物的影响[7-8].植物的生长与土壤微生物密切相关,土壤微生物群落代表了迄今为止世界上已知最大的生物多样性库[9].根际是受根系分泌物影响的狭窄土壤微生态位,每克根可含有多达1011个微生物菌体[10]及3万多种原核生物[11].越来越多的证据表明,这种根际微生物群落在决定相关植物健康和功能等方面具有重要作用,涉及植物根系内生菌群落及其遗传因素和多种环境因子综合的交互性作用[9-14].如根际土壤微生物群落对植物抗病害方面的影响最为显著,大多数在土壤中传播的病原体需要在根际腐生生长才能够接触到宿主或在宿主周围达到足够数量,进而有效地感染宿主并脱离根际微域,而病原体能否成功感染宿主植物,受根际土壤微生物群落的显著影响[15].还有研究指出,根际微生物群落对宿主植物的免疫系统具有调节作用[16].而植物内生微生物是指寄生在植物组织内至少一段时间的细菌、古生菌、真菌或病毒群落,对寄主无任何不利影响[17].它们在促进植物的生长发育、营养储备和抵御生物或非生物胁迫(如疾病、虫害、高温、盐或干旱)等方面起着关键作用[18-20].豆科植物与根瘤菌的共生关系通过生物固氮和协同进化对于改善土壤质地和肥力具有巨大潜力[21].Bulgarelli等[22]发现,一些植物内生菌是从根际土向植物根系内迁移而来的,并主要受到根际土壤因子的影响. ...
Microbial inoculants and organic amendment improves plant establishment and soil rehabilitation under semiarid conditions
1
2014
... 在生态环境整体恶劣的荒漠地区,植物群落的定植生长能够起到防风固沙的功效,尤以植物发达延伸生长的根系能够固持和改良土壤[5].土壤作为植物生长的载体,不仅为植株生长提供养分支持,同时也在植物固定外来碳源的作用下,改良提升自身的理化和生物特性[6],而这其中土壤微生物发挥着重要的作用.荒漠植物群落的适应性很大程度上受土壤微生物的影响[7-8].植物的生长与土壤微生物密切相关,土壤微生物群落代表了迄今为止世界上已知最大的生物多样性库[9].根际是受根系分泌物影响的狭窄土壤微生态位,每克根可含有多达1011个微生物菌体[10]及3万多种原核生物[11].越来越多的证据表明,这种根际微生物群落在决定相关植物健康和功能等方面具有重要作用,涉及植物根系内生菌群落及其遗传因素和多种环境因子综合的交互性作用[9-14].如根际土壤微生物群落对植物抗病害方面的影响最为显著,大多数在土壤中传播的病原体需要在根际腐生生长才能够接触到宿主或在宿主周围达到足够数量,进而有效地感染宿主并脱离根际微域,而病原体能否成功感染宿主植物,受根际土壤微生物群落的显著影响[15].还有研究指出,根际微生物群落对宿主植物的免疫系统具有调节作用[16].而植物内生微生物是指寄生在植物组织内至少一段时间的细菌、古生菌、真菌或病毒群落,对寄主无任何不利影响[17].它们在促进植物的生长发育、营养储备和抵御生物或非生物胁迫(如疾病、虫害、高温、盐或干旱)等方面起着关键作用[18-20].豆科植物与根瘤菌的共生关系通过生物固氮和协同进化对于改善土壤质地和肥力具有巨大潜力[21].Bulgarelli等[22]发现,一些植物内生菌是从根际土向植物根系内迁移而来的,并主要受到根际土壤因子的影响. ...
The rhizosphere microbiome and plant health
4
2012
... 在生态环境整体恶劣的荒漠地区,植物群落的定植生长能够起到防风固沙的功效,尤以植物发达延伸生长的根系能够固持和改良土壤[5].土壤作为植物生长的载体,不仅为植株生长提供养分支持,同时也在植物固定外来碳源的作用下,改良提升自身的理化和生物特性[6],而这其中土壤微生物发挥着重要的作用.荒漠植物群落的适应性很大程度上受土壤微生物的影响[7-8].植物的生长与土壤微生物密切相关,土壤微生物群落代表了迄今为止世界上已知最大的生物多样性库[9].根际是受根系分泌物影响的狭窄土壤微生态位,每克根可含有多达1011个微生物菌体[10]及3万多种原核生物[11].越来越多的证据表明,这种根际微生物群落在决定相关植物健康和功能等方面具有重要作用,涉及植物根系内生菌群落及其遗传因素和多种环境因子综合的交互性作用[9-14].如根际土壤微生物群落对植物抗病害方面的影响最为显著,大多数在土壤中传播的病原体需要在根际腐生生长才能够接触到宿主或在宿主周围达到足够数量,进而有效地感染宿主并脱离根际微域,而病原体能否成功感染宿主植物,受根际土壤微生物群落的显著影响[15].还有研究指出,根际微生物群落对宿主植物的免疫系统具有调节作用[16].而植物内生微生物是指寄生在植物组织内至少一段时间的细菌、古生菌、真菌或病毒群落,对寄主无任何不利影响[17].它们在促进植物的生长发育、营养储备和抵御生物或非生物胁迫(如疾病、虫害、高温、盐或干旱)等方面起着关键作用[18-20].豆科植物与根瘤菌的共生关系通过生物固氮和协同进化对于改善土壤质地和肥力具有巨大潜力[21].Bulgarelli等[22]发现,一些植物内生菌是从根际土向植物根系内迁移而来的,并主要受到根际土壤因子的影响. ...
... [9-14].如根际土壤微生物群落对植物抗病害方面的影响最为显著,大多数在土壤中传播的病原体需要在根际腐生生长才能够接触到宿主或在宿主周围达到足够数量,进而有效地感染宿主并脱离根际微域,而病原体能否成功感染宿主植物,受根际土壤微生物群落的显著影响[15].还有研究指出,根际微生物群落对宿主植物的免疫系统具有调节作用[16].而植物内生微生物是指寄生在植物组织内至少一段时间的细菌、古生菌、真菌或病毒群落,对寄主无任何不利影响[17].它们在促进植物的生长发育、营养储备和抵御生物或非生物胁迫(如疾病、虫害、高温、盐或干旱)等方面起着关键作用[18-20].豆科植物与根瘤菌的共生关系通过生物固氮和协同进化对于改善土壤质地和肥力具有巨大潜力[21].Bulgarelli等[22]发现,一些植物内生菌是从根际土向植物根系内迁移而来的,并主要受到根际土壤因子的影响. ...
... 根际土中微生物群落的基因组比植物丰富,也被称为植物的第二基因组[9].根际微生物在调节植物个体的健康、适应性和群落生产力等方面都发挥着难以替代的作用[9,23-26],而植物群落又通过影响土壤养分循环等生态过程影响土壤微生物群落[6].此外,部分根际微生物可以进入植物根系组织形成植物内生菌群落,其群落结构组成与根际土壤微生物群落又有显著的差异性[27-28],这种细菌群落多样性和结构组成在根系组织内和根际土之间的显著差异现象被定义为“根际效应”[29].Sun等[30]发现,毛乌素沙地旱生灌丛下和灌丛间的土壤细菌群落也出现在了细菌群落结构组成显著差异的现象.锦鸡儿属植物对荒漠环境的适应能力和生态恢复作用始终都受到高度的关注[5,31-32].基于以上的研究结论,本研究的主旨设定为:①荒漠豆科锦鸡儿属灌丛下的4个根系微域(根系、根际土、根区土、灌丛间空白土)存在细菌群落多样性和结构组成的层级差异性变化;②探究荒漠豆科锦鸡儿属植物灌丛下根系微域细菌群落结构的层级变异性,及其发生变异的成因. ...
... [9,23-26],而植物群落又通过影响土壤养分循环等生态过程影响土壤微生物群落[6].此外,部分根际微生物可以进入植物根系组织形成植物内生菌群落,其群落结构组成与根际土壤微生物群落又有显著的差异性[27-28],这种细菌群落多样性和结构组成在根系组织内和根际土之间的显著差异现象被定义为“根际效应”[29].Sun等[30]发现,毛乌素沙地旱生灌丛下和灌丛间的土壤细菌群落也出现在了细菌群落结构组成显著差异的现象.锦鸡儿属植物对荒漠环境的适应能力和生态恢复作用始终都受到高度的关注[5,31-32].基于以上的研究结论,本研究的主旨设定为:①荒漠豆科锦鸡儿属灌丛下的4个根系微域(根系、根际土、根区土、灌丛间空白土)存在细菌群落多样性和结构组成的层级差异性变化;②探究荒漠豆科锦鸡儿属植物灌丛下根系微域细菌群落结构的层级变异性,及其发生变异的成因. ...
High incidence of plant growth-stimulating bacteria associated with the rhizosphere of wheat grown on salinated soil in Uzbekistan
1
2008
... 在生态环境整体恶劣的荒漠地区,植物群落的定植生长能够起到防风固沙的功效,尤以植物发达延伸生长的根系能够固持和改良土壤[5].土壤作为植物生长的载体,不仅为植株生长提供养分支持,同时也在植物固定外来碳源的作用下,改良提升自身的理化和生物特性[6],而这其中土壤微生物发挥着重要的作用.荒漠植物群落的适应性很大程度上受土壤微生物的影响[7-8].植物的生长与土壤微生物密切相关,土壤微生物群落代表了迄今为止世界上已知最大的生物多样性库[9].根际是受根系分泌物影响的狭窄土壤微生态位,每克根可含有多达1011个微生物菌体[10]及3万多种原核生物[11].越来越多的证据表明,这种根际微生物群落在决定相关植物健康和功能等方面具有重要作用,涉及植物根系内生菌群落及其遗传因素和多种环境因子综合的交互性作用[9-14].如根际土壤微生物群落对植物抗病害方面的影响最为显著,大多数在土壤中传播的病原体需要在根际腐生生长才能够接触到宿主或在宿主周围达到足够数量,进而有效地感染宿主并脱离根际微域,而病原体能否成功感染宿主植物,受根际土壤微生物群落的显著影响[15].还有研究指出,根际微生物群落对宿主植物的免疫系统具有调节作用[16].而植物内生微生物是指寄生在植物组织内至少一段时间的细菌、古生菌、真菌或病毒群落,对寄主无任何不利影响[17].它们在促进植物的生长发育、营养储备和抵御生物或非生物胁迫(如疾病、虫害、高温、盐或干旱)等方面起着关键作用[18-20].豆科植物与根瘤菌的共生关系通过生物固氮和协同进化对于改善土壤质地和肥力具有巨大潜力[21].Bulgarelli等[22]发现,一些植物内生菌是从根际土向植物根系内迁移而来的,并主要受到根际土壤因子的影响. ...
Deciphering the rhizosphere microbiome for disease-suppressive bacteria
1
2011
... 在生态环境整体恶劣的荒漠地区,植物群落的定植生长能够起到防风固沙的功效,尤以植物发达延伸生长的根系能够固持和改良土壤[5].土壤作为植物生长的载体,不仅为植株生长提供养分支持,同时也在植物固定外来碳源的作用下,改良提升自身的理化和生物特性[6],而这其中土壤微生物发挥着重要的作用.荒漠植物群落的适应性很大程度上受土壤微生物的影响[7-8].植物的生长与土壤微生物密切相关,土壤微生物群落代表了迄今为止世界上已知最大的生物多样性库[9].根际是受根系分泌物影响的狭窄土壤微生态位,每克根可含有多达1011个微生物菌体[10]及3万多种原核生物[11].越来越多的证据表明,这种根际微生物群落在决定相关植物健康和功能等方面具有重要作用,涉及植物根系内生菌群落及其遗传因素和多种环境因子综合的交互性作用[9-14].如根际土壤微生物群落对植物抗病害方面的影响最为显著,大多数在土壤中传播的病原体需要在根际腐生生长才能够接触到宿主或在宿主周围达到足够数量,进而有效地感染宿主并脱离根际微域,而病原体能否成功感染宿主植物,受根际土壤微生物群落的显著影响[15].还有研究指出,根际微生物群落对宿主植物的免疫系统具有调节作用[16].而植物内生微生物是指寄生在植物组织内至少一段时间的细菌、古生菌、真菌或病毒群落,对寄主无任何不利影响[17].它们在促进植物的生长发育、营养储备和抵御生物或非生物胁迫(如疾病、虫害、高温、盐或干旱)等方面起着关键作用[18-20].豆科植物与根瘤菌的共生关系通过生物固氮和协同进化对于改善土壤质地和肥力具有巨大潜力[21].Bulgarelli等[22]发现,一些植物内生菌是从根际土向植物根系内迁移而来的,并主要受到根际土壤因子的影响. ...
Structural variability and niche differentiation in the rhizosphere and endosphere bacterial microbiome of field-grown poplar trees
3
2017
... 综上所述,这种与寄主植物相关的优势菌群在各根系微域逐级向根系富集的趋势或许并不只是由寄主植物对有益细菌的筛选作用所主导的,还可能是由某些细菌对适宜生态位的投机性迁移、定殖引起的[12]. ...
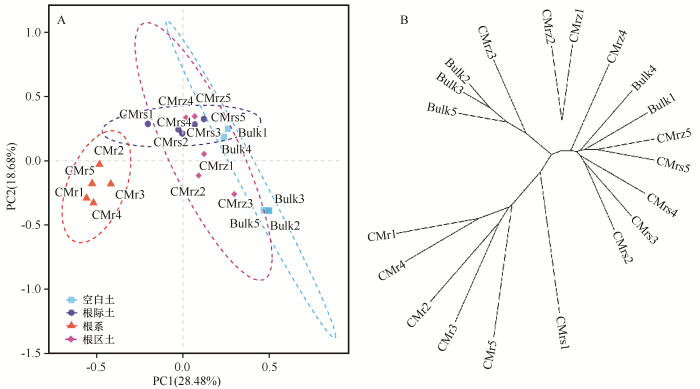
... 荒漠豆科锦鸡儿属植物4个根系微域的样本间Alpha和Beta多样性都存在显著的差异性(P<0.05).荒漠豆科锦鸡儿属植物通过根系微域对灌丛下土壤细菌群落中特定的优势菌群具有筛选富集作用.Beckers等[12]在毛白杨的根系内和根际土生态位的研究中也得到了类似的根系微细域菌群落结构差异性结果,并认为这可能是由于灌丛下的细菌群落是随着根系分布而迁移定殖不均匀造成的.我们的数据表明,植株对根系内定殖的大量内生菌中特定的细菌微生物群组的筛选富集作用是根系内生菌群落发生高度变异性的主要原因.现有的研究表明,向植物根系对特定细菌群组的筛选富集主要通过以下两种途径:①根系分泌物,尤其是植物根际部位分泌含碳代谢产物[45-46];②植物通过化感作用释放化感物质,对土壤细菌微生物产生的化学诱导作用[47].虽然根系分泌物等衍生的营养物质和趋化诱导在根际微域这个生态位广泛存在,但与植物相关的特定有益细菌则需要通过激烈的养分竞争才能成功地迁移并定殖于根系内[48].综上所述,内生菌迁移并定殖于根系内形成一个结构组成相对稳定的群落似乎经历了一个高度变异的过程,这从我们的Alpha多样性(表2)、主成分分析(图1)、根系微域间优势菌群筛选富集分析(图3)、ANOSIM细菌群落结构相似性分析(表3)中都可以得出这样的结果. ...
... 此外,我们发现在根系微域间还存在大量相对丰度差异性变化的菌物,其绝大多数都被植物根系筛选富集,并且这种寄主植物内生菌群落组成,优势菌属在根系微域间大量重叠的现象说明(图3),内生能力(有效的筛选、富集、定殖)和植物根系微域(营养有效性/变异性、生境适宜性等)都是为前述涉及的特定优势菌群保留的[12]. ...
Dynamics of the bacterial community structure in the rhizosphere of a maize cultivar
2014
Endophytic communities of transgenic poplar were determined by the environment and niche rather than by transgenic events
1
2019
... 在生态环境整体恶劣的荒漠地区,植物群落的定植生长能够起到防风固沙的功效,尤以植物发达延伸生长的根系能够固持和改良土壤[5].土壤作为植物生长的载体,不仅为植株生长提供养分支持,同时也在植物固定外来碳源的作用下,改良提升自身的理化和生物特性[6],而这其中土壤微生物发挥着重要的作用.荒漠植物群落的适应性很大程度上受土壤微生物的影响[7-8].植物的生长与土壤微生物密切相关,土壤微生物群落代表了迄今为止世界上已知最大的生物多样性库[9].根际是受根系分泌物影响的狭窄土壤微生态位,每克根可含有多达1011个微生物菌体[10]及3万多种原核生物[11].越来越多的证据表明,这种根际微生物群落在决定相关植物健康和功能等方面具有重要作用,涉及植物根系内生菌群落及其遗传因素和多种环境因子综合的交互性作用[9-14].如根际土壤微生物群落对植物抗病害方面的影响最为显著,大多数在土壤中传播的病原体需要在根际腐生生长才能够接触到宿主或在宿主周围达到足够数量,进而有效地感染宿主并脱离根际微域,而病原体能否成功感染宿主植物,受根际土壤微生物群落的显著影响[15].还有研究指出,根际微生物群落对宿主植物的免疫系统具有调节作用[16].而植物内生微生物是指寄生在植物组织内至少一段时间的细菌、古生菌、真菌或病毒群落,对寄主无任何不利影响[17].它们在促进植物的生长发育、营养储备和抵御生物或非生物胁迫(如疾病、虫害、高温、盐或干旱)等方面起着关键作用[18-20].豆科植物与根瘤菌的共生关系通过生物固氮和协同进化对于改善土壤质地和肥力具有巨大潜力[21].Bulgarelli等[22]发现,一些植物内生菌是从根际土向植物根系内迁移而来的,并主要受到根际土壤因子的影响. ...
Biocontrol within the context of soil microbial communities:a substrate-dependent phenomenon
1
1999
... 在生态环境整体恶劣的荒漠地区,植物群落的定植生长能够起到防风固沙的功效,尤以植物发达延伸生长的根系能够固持和改良土壤[5].土壤作为植物生长的载体,不仅为植株生长提供养分支持,同时也在植物固定外来碳源的作用下,改良提升自身的理化和生物特性[6],而这其中土壤微生物发挥着重要的作用.荒漠植物群落的适应性很大程度上受土壤微生物的影响[7-8].植物的生长与土壤微生物密切相关,土壤微生物群落代表了迄今为止世界上已知最大的生物多样性库[9].根际是受根系分泌物影响的狭窄土壤微生态位,每克根可含有多达1011个微生物菌体[10]及3万多种原核生物[11].越来越多的证据表明,这种根际微生物群落在决定相关植物健康和功能等方面具有重要作用,涉及植物根系内生菌群落及其遗传因素和多种环境因子综合的交互性作用[9-14].如根际土壤微生物群落对植物抗病害方面的影响最为显著,大多数在土壤中传播的病原体需要在根际腐生生长才能够接触到宿主或在宿主周围达到足够数量,进而有效地感染宿主并脱离根际微域,而病原体能否成功感染宿主植物,受根际土壤微生物群落的显著影响[15].还有研究指出,根际微生物群落对宿主植物的免疫系统具有调节作用[16].而植物内生微生物是指寄生在植物组织内至少一段时间的细菌、古生菌、真菌或病毒群落,对寄主无任何不利影响[17].它们在促进植物的生长发育、营养储备和抵御生物或非生物胁迫(如疾病、虫害、高温、盐或干旱)等方面起着关键作用[18-20].豆科植物与根瘤菌的共生关系通过生物固氮和协同进化对于改善土壤质地和肥力具有巨大潜力[21].Bulgarelli等[22]发现,一些植物内生菌是从根际土向植物根系内迁移而来的,并主要受到根际土壤因子的影响. ...
Innate immune responses activated in Arabidopsis roots by microbe-associated molecular patterns
1
2010
... 在生态环境整体恶劣的荒漠地区,植物群落的定植生长能够起到防风固沙的功效,尤以植物发达延伸生长的根系能够固持和改良土壤[5].土壤作为植物生长的载体,不仅为植株生长提供养分支持,同时也在植物固定外来碳源的作用下,改良提升自身的理化和生物特性[6],而这其中土壤微生物发挥着重要的作用.荒漠植物群落的适应性很大程度上受土壤微生物的影响[7-8].植物的生长与土壤微生物密切相关,土壤微生物群落代表了迄今为止世界上已知最大的生物多样性库[9].根际是受根系分泌物影响的狭窄土壤微生态位,每克根可含有多达1011个微生物菌体[10]及3万多种原核生物[11].越来越多的证据表明,这种根际微生物群落在决定相关植物健康和功能等方面具有重要作用,涉及植物根系内生菌群落及其遗传因素和多种环境因子综合的交互性作用[9-14].如根际土壤微生物群落对植物抗病害方面的影响最为显著,大多数在土壤中传播的病原体需要在根际腐生生长才能够接触到宿主或在宿主周围达到足够数量,进而有效地感染宿主并脱离根际微域,而病原体能否成功感染宿主植物,受根际土壤微生物群落的显著影响[15].还有研究指出,根际微生物群落对宿主植物的免疫系统具有调节作用[16].而植物内生微生物是指寄生在植物组织内至少一段时间的细菌、古生菌、真菌或病毒群落,对寄主无任何不利影响[17].它们在促进植物的生长发育、营养储备和抵御生物或非生物胁迫(如疾病、虫害、高温、盐或干旱)等方面起着关键作用[18-20].豆科植物与根瘤菌的共生关系通过生物固氮和协同进化对于改善土壤质地和肥力具有巨大潜力[21].Bulgarelli等[22]发现,一些植物内生菌是从根际土向植物根系内迁移而来的,并主要受到根际土壤因子的影响. ...
Endophytic bacteria in agricultural crops
1
2011
... 在生态环境整体恶劣的荒漠地区,植物群落的定植生长能够起到防风固沙的功效,尤以植物发达延伸生长的根系能够固持和改良土壤[5].土壤作为植物生长的载体,不仅为植株生长提供养分支持,同时也在植物固定外来碳源的作用下,改良提升自身的理化和生物特性[6],而这其中土壤微生物发挥着重要的作用.荒漠植物群落的适应性很大程度上受土壤微生物的影响[7-8].植物的生长与土壤微生物密切相关,土壤微生物群落代表了迄今为止世界上已知最大的生物多样性库[9].根际是受根系分泌物影响的狭窄土壤微生态位,每克根可含有多达1011个微生物菌体[10]及3万多种原核生物[11].越来越多的证据表明,这种根际微生物群落在决定相关植物健康和功能等方面具有重要作用,涉及植物根系内生菌群落及其遗传因素和多种环境因子综合的交互性作用[9-14].如根际土壤微生物群落对植物抗病害方面的影响最为显著,大多数在土壤中传播的病原体需要在根际腐生生长才能够接触到宿主或在宿主周围达到足够数量,进而有效地感染宿主并脱离根际微域,而病原体能否成功感染宿主植物,受根际土壤微生物群落的显著影响[15].还有研究指出,根际微生物群落对宿主植物的免疫系统具有调节作用[16].而植物内生微生物是指寄生在植物组织内至少一段时间的细菌、古生菌、真菌或病毒群落,对寄主无任何不利影响[17].它们在促进植物的生长发育、营养储备和抵御生物或非生物胁迫(如疾病、虫害、高温、盐或干旱)等方面起着关键作用[18-20].豆科植物与根瘤菌的共生关系通过生物固氮和协同进化对于改善土壤质地和肥力具有巨大潜力[21].Bulgarelli等[22]发现,一些植物内生菌是从根际土向植物根系内迁移而来的,并主要受到根际土壤因子的影响. ...
Properties of bacterial endophytes and their proposed role in plant growth
1
2008
... 在生态环境整体恶劣的荒漠地区,植物群落的定植生长能够起到防风固沙的功效,尤以植物发达延伸生长的根系能够固持和改良土壤[5].土壤作为植物生长的载体,不仅为植株生长提供养分支持,同时也在植物固定外来碳源的作用下,改良提升自身的理化和生物特性[6],而这其中土壤微生物发挥着重要的作用.荒漠植物群落的适应性很大程度上受土壤微生物的影响[7-8].植物的生长与土壤微生物密切相关,土壤微生物群落代表了迄今为止世界上已知最大的生物多样性库[9].根际是受根系分泌物影响的狭窄土壤微生态位,每克根可含有多达1011个微生物菌体[10]及3万多种原核生物[11].越来越多的证据表明,这种根际微生物群落在决定相关植物健康和功能等方面具有重要作用,涉及植物根系内生菌群落及其遗传因素和多种环境因子综合的交互性作用[9-14].如根际土壤微生物群落对植物抗病害方面的影响最为显著,大多数在土壤中传播的病原体需要在根际腐生生长才能够接触到宿主或在宿主周围达到足够数量,进而有效地感染宿主并脱离根际微域,而病原体能否成功感染宿主植物,受根际土壤微生物群落的显著影响[15].还有研究指出,根际微生物群落对宿主植物的免疫系统具有调节作用[16].而植物内生微生物是指寄生在植物组织内至少一段时间的细菌、古生菌、真菌或病毒群落,对寄主无任何不利影响[17].它们在促进植物的生长发育、营养储备和抵御生物或非生物胁迫(如疾病、虫害、高温、盐或干旱)等方面起着关键作用[18-20].豆科植物与根瘤菌的共生关系通过生物固氮和协同进化对于改善土壤质地和肥力具有巨大潜力[21].Bulgarelli等[22]发现,一些植物内生菌是从根际土向植物根系内迁移而来的,并主要受到根际土壤因子的影响. ...
Genetically modified Btmaize lines containing cry3Bb1,cry1A105 or cry1Ab2 do not affect the structure and functioning of root-associated endophyte communities
2012
Increased drought stress resilience of maize through endophytic colonization by Burkholderiaphytofirmans PsJN and Enterobacter sp.FD17
1
2014
... 在生态环境整体恶劣的荒漠地区,植物群落的定植生长能够起到防风固沙的功效,尤以植物发达延伸生长的根系能够固持和改良土壤[5].土壤作为植物生长的载体,不仅为植株生长提供养分支持,同时也在植物固定外来碳源的作用下,改良提升自身的理化和生物特性[6],而这其中土壤微生物发挥着重要的作用.荒漠植物群落的适应性很大程度上受土壤微生物的影响[7-8].植物的生长与土壤微生物密切相关,土壤微生物群落代表了迄今为止世界上已知最大的生物多样性库[9].根际是受根系分泌物影响的狭窄土壤微生态位,每克根可含有多达1011个微生物菌体[10]及3万多种原核生物[11].越来越多的证据表明,这种根际微生物群落在决定相关植物健康和功能等方面具有重要作用,涉及植物根系内生菌群落及其遗传因素和多种环境因子综合的交互性作用[9-14].如根际土壤微生物群落对植物抗病害方面的影响最为显著,大多数在土壤中传播的病原体需要在根际腐生生长才能够接触到宿主或在宿主周围达到足够数量,进而有效地感染宿主并脱离根际微域,而病原体能否成功感染宿主植物,受根际土壤微生物群落的显著影响[15].还有研究指出,根际微生物群落对宿主植物的免疫系统具有调节作用[16].而植物内生微生物是指寄生在植物组织内至少一段时间的细菌、古生菌、真菌或病毒群落,对寄主无任何不利影响[17].它们在促进植物的生长发育、营养储备和抵御生物或非生物胁迫(如疾病、虫害、高温、盐或干旱)等方面起着关键作用[18-20].豆科植物与根瘤菌的共生关系通过生物固氮和协同进化对于改善土壤质地和肥力具有巨大潜力[21].Bulgarelli等[22]发现,一些植物内生菌是从根际土向植物根系内迁移而来的,并主要受到根际土壤因子的影响. ...
Microbial hub Taxa link host and abiotic factors to plant microbiome variation
1
2016
... 在生态环境整体恶劣的荒漠地区,植物群落的定植生长能够起到防风固沙的功效,尤以植物发达延伸生长的根系能够固持和改良土壤[5].土壤作为植物生长的载体,不仅为植株生长提供养分支持,同时也在植物固定外来碳源的作用下,改良提升自身的理化和生物特性[6],而这其中土壤微生物发挥着重要的作用.荒漠植物群落的适应性很大程度上受土壤微生物的影响[7-8].植物的生长与土壤微生物密切相关,土壤微生物群落代表了迄今为止世界上已知最大的生物多样性库[9].根际是受根系分泌物影响的狭窄土壤微生态位,每克根可含有多达1011个微生物菌体[10]及3万多种原核生物[11].越来越多的证据表明,这种根际微生物群落在决定相关植物健康和功能等方面具有重要作用,涉及植物根系内生菌群落及其遗传因素和多种环境因子综合的交互性作用[9-14].如根际土壤微生物群落对植物抗病害方面的影响最为显著,大多数在土壤中传播的病原体需要在根际腐生生长才能够接触到宿主或在宿主周围达到足够数量,进而有效地感染宿主并脱离根际微域,而病原体能否成功感染宿主植物,受根际土壤微生物群落的显著影响[15].还有研究指出,根际微生物群落对宿主植物的免疫系统具有调节作用[16].而植物内生微生物是指寄生在植物组织内至少一段时间的细菌、古生菌、真菌或病毒群落,对寄主无任何不利影响[17].它们在促进植物的生长发育、营养储备和抵御生物或非生物胁迫(如疾病、虫害、高温、盐或干旱)等方面起着关键作用[18-20].豆科植物与根瘤菌的共生关系通过生物固氮和协同进化对于改善土壤质地和肥力具有巨大潜力[21].Bulgarelli等[22]发现,一些植物内生菌是从根际土向植物根系内迁移而来的,并主要受到根际土壤因子的影响. ...
Structure and function of the bacterial root microbiota in wild and domesticated barley
1
2015
... 在生态环境整体恶劣的荒漠地区,植物群落的定植生长能够起到防风固沙的功效,尤以植物发达延伸生长的根系能够固持和改良土壤[5].土壤作为植物生长的载体,不仅为植株生长提供养分支持,同时也在植物固定外来碳源的作用下,改良提升自身的理化和生物特性[6],而这其中土壤微生物发挥着重要的作用.荒漠植物群落的适应性很大程度上受土壤微生物的影响[7-8].植物的生长与土壤微生物密切相关,土壤微生物群落代表了迄今为止世界上已知最大的生物多样性库[9].根际是受根系分泌物影响的狭窄土壤微生态位,每克根可含有多达1011个微生物菌体[10]及3万多种原核生物[11].越来越多的证据表明,这种根际微生物群落在决定相关植物健康和功能等方面具有重要作用,涉及植物根系内生菌群落及其遗传因素和多种环境因子综合的交互性作用[9-14].如根际土壤微生物群落对植物抗病害方面的影响最为显著,大多数在土壤中传播的病原体需要在根际腐生生长才能够接触到宿主或在宿主周围达到足够数量,进而有效地感染宿主并脱离根际微域,而病原体能否成功感染宿主植物,受根际土壤微生物群落的显著影响[15].还有研究指出,根际微生物群落对宿主植物的免疫系统具有调节作用[16].而植物内生微生物是指寄生在植物组织内至少一段时间的细菌、古生菌、真菌或病毒群落,对寄主无任何不利影响[17].它们在促进植物的生长发育、营养储备和抵御生物或非生物胁迫(如疾病、虫害、高温、盐或干旱)等方面起着关键作用[18-20].豆科植物与根瘤菌的共生关系通过生物固氮和协同进化对于改善土壤质地和肥力具有巨大潜力[21].Bulgarelli等[22]发现,一些植物内生菌是从根际土向植物根系内迁移而来的,并主要受到根际土壤因子的影响. ...
Host genetic control of symbiosis in soybean (Glycine max L.)
1
1996
... 根际土中微生物群落的基因组比植物丰富,也被称为植物的第二基因组[9].根际微生物在调节植物个体的健康、适应性和群落生产力等方面都发挥着难以替代的作用[9,23-26],而植物群落又通过影响土壤养分循环等生态过程影响土壤微生物群落[6].此外,部分根际微生物可以进入植物根系组织形成植物内生菌群落,其群落结构组成与根际土壤微生物群落又有显著的差异性[27-28],这种细菌群落多样性和结构组成在根系组织内和根际土之间的显著差异现象被定义为“根际效应”[29].Sun等[30]发现,毛乌素沙地旱生灌丛下和灌丛间的土壤细菌群落也出现在了细菌群落结构组成显著差异的现象.锦鸡儿属植物对荒漠环境的适应能力和生态恢复作用始终都受到高度的关注[5,31-32].基于以上的研究结论,本研究的主旨设定为:①荒漠豆科锦鸡儿属灌丛下的4个根系微域(根系、根际土、根区土、灌丛间空白土)存在细菌群落多样性和结构组成的层级差异性变化;②探究荒漠豆科锦鸡儿属植物灌丛下根系微域细菌群落结构的层级变异性,及其发生变异的成因. ...
Unraveling the plant microbiome:looking back and future perspectives
2014
Partner diversity and identity impacts on plant productivity in Acacia-rhizobial interactions
2015
Larger plants promote a greater diversity of symbiotic nitrogen fixing soil bacteria associated with an Australian endemic legume
1
2018
... 根际土中微生物群落的基因组比植物丰富,也被称为植物的第二基因组[9].根际微生物在调节植物个体的健康、适应性和群落生产力等方面都发挥着难以替代的作用[9,23-26],而植物群落又通过影响土壤养分循环等生态过程影响土壤微生物群落[6].此外,部分根际微生物可以进入植物根系组织形成植物内生菌群落,其群落结构组成与根际土壤微生物群落又有显著的差异性[27-28],这种细菌群落多样性和结构组成在根系组织内和根际土之间的显著差异现象被定义为“根际效应”[29].Sun等[30]发现,毛乌素沙地旱生灌丛下和灌丛间的土壤细菌群落也出现在了细菌群落结构组成显著差异的现象.锦鸡儿属植物对荒漠环境的适应能力和生态恢复作用始终都受到高度的关注[5,31-32].基于以上的研究结论,本研究的主旨设定为:①荒漠豆科锦鸡儿属灌丛下的4个根系微域(根系、根际土、根区土、灌丛间空白土)存在细菌群落多样性和结构组成的层级差异性变化;②探究荒漠豆科锦鸡儿属植物灌丛下根系微域细菌群落结构的层级变异性,及其发生变异的成因. ...
Distinct microbial communities within the endosphere and rhizosphere of Populus deltoides roots across contrasting soil types
1
2011
... 根际土中微生物群落的基因组比植物丰富,也被称为植物的第二基因组[9].根际微生物在调节植物个体的健康、适应性和群落生产力等方面都发挥着难以替代的作用[9,23-26],而植物群落又通过影响土壤养分循环等生态过程影响土壤微生物群落[6].此外,部分根际微生物可以进入植物根系组织形成植物内生菌群落,其群落结构组成与根际土壤微生物群落又有显著的差异性[27-28],这种细菌群落多样性和结构组成在根系组织内和根际土之间的显著差异现象被定义为“根际效应”[29].Sun等[30]发现,毛乌素沙地旱生灌丛下和灌丛间的土壤细菌群落也出现在了细菌群落结构组成显著差异的现象.锦鸡儿属植物对荒漠环境的适应能力和生态恢复作用始终都受到高度的关注[5,31-32].基于以上的研究结论,本研究的主旨设定为:①荒漠豆科锦鸡儿属灌丛下的4个根系微域(根系、根际土、根区土、灌丛间空白土)存在细菌群落多样性和结构组成的层级差异性变化;②探究荒漠豆科锦鸡儿属植物灌丛下根系微域细菌群落结构的层级变异性,及其发生变异的成因. ...
Defining the core Arabidopsis thaliana root microbiome
1
2012
... 根际土中微生物群落的基因组比植物丰富,也被称为植物的第二基因组[9].根际微生物在调节植物个体的健康、适应性和群落生产力等方面都发挥着难以替代的作用[9,23-26],而植物群落又通过影响土壤养分循环等生态过程影响土壤微生物群落[6].此外,部分根际微生物可以进入植物根系组织形成植物内生菌群落,其群落结构组成与根际土壤微生物群落又有显著的差异性[27-28],这种细菌群落多样性和结构组成在根系组织内和根际土之间的显著差异现象被定义为“根际效应”[29].Sun等[30]发现,毛乌素沙地旱生灌丛下和灌丛间的土壤细菌群落也出现在了细菌群落结构组成显著差异的现象.锦鸡儿属植物对荒漠环境的适应能力和生态恢复作用始终都受到高度的关注[5,31-32].基于以上的研究结论,本研究的主旨设定为:①荒漠豆科锦鸡儿属灌丛下的4个根系微域(根系、根际土、根区土、灌丛间空白土)存在细菌群落多样性和结构组成的层级差异性变化;②探究荒漠豆科锦鸡儿属植物灌丛下根系微域细菌群落结构的层级变异性,及其发生变异的成因. ...
Lorenz Hiltner,a pioneer in rhizosphere microbial ecology and bacteriology research
1
2008
... 根际土中微生物群落的基因组比植物丰富,也被称为植物的第二基因组[9].根际微生物在调节植物个体的健康、适应性和群落生产力等方面都发挥着难以替代的作用[9,23-26],而植物群落又通过影响土壤养分循环等生态过程影响土壤微生物群落[6].此外,部分根际微生物可以进入植物根系组织形成植物内生菌群落,其群落结构组成与根际土壤微生物群落又有显著的差异性[27-28],这种细菌群落多样性和结构组成在根系组织内和根际土之间的显著差异现象被定义为“根际效应”[29].Sun等[30]发现,毛乌素沙地旱生灌丛下和灌丛间的土壤细菌群落也出现在了细菌群落结构组成显著差异的现象.锦鸡儿属植物对荒漠环境的适应能力和生态恢复作用始终都受到高度的关注[5,31-32].基于以上的研究结论,本研究的主旨设定为:①荒漠豆科锦鸡儿属灌丛下的4个根系微域(根系、根际土、根区土、灌丛间空白土)存在细菌群落多样性和结构组成的层级差异性变化;②探究荒漠豆科锦鸡儿属植物灌丛下根系微域细菌群落结构的层级变异性,及其发生变异的成因. ...
Effects of xeric shrubs on soil microbial communities in a desert in northern china
1
2017
... 根际土中微生物群落的基因组比植物丰富,也被称为植物的第二基因组[9].根际微生物在调节植物个体的健康、适应性和群落生产力等方面都发挥着难以替代的作用[9,23-26],而植物群落又通过影响土壤养分循环等生态过程影响土壤微生物群落[6].此外,部分根际微生物可以进入植物根系组织形成植物内生菌群落,其群落结构组成与根际土壤微生物群落又有显著的差异性[27-28],这种细菌群落多样性和结构组成在根系组织内和根际土之间的显著差异现象被定义为“根际效应”[29].Sun等[30]发现,毛乌素沙地旱生灌丛下和灌丛间的土壤细菌群落也出现在了细菌群落结构组成显著差异的现象.锦鸡儿属植物对荒漠环境的适应能力和生态恢复作用始终都受到高度的关注[5,31-32].基于以上的研究结论,本研究的主旨设定为:①荒漠豆科锦鸡儿属灌丛下的4个根系微域(根系、根际土、根区土、灌丛间空白土)存在细菌群落多样性和结构组成的层级差异性变化;②探究荒漠豆科锦鸡儿属植物灌丛下根系微域细菌群落结构的层级变异性,及其发生变异的成因. ...
How the plant temperature links to the air temperature in the desert plant Artemisia ordosica
2
2015
... 根际土中微生物群落的基因组比植物丰富,也被称为植物的第二基因组[9].根际微生物在调节植物个体的健康、适应性和群落生产力等方面都发挥着难以替代的作用[9,23-26],而植物群落又通过影响土壤养分循环等生态过程影响土壤微生物群落[6].此外,部分根际微生物可以进入植物根系组织形成植物内生菌群落,其群落结构组成与根际土壤微生物群落又有显著的差异性[27-28],这种细菌群落多样性和结构组成在根系组织内和根际土之间的显著差异现象被定义为“根际效应”[29].Sun等[30]发现,毛乌素沙地旱生灌丛下和灌丛间的土壤细菌群落也出现在了细菌群落结构组成显著差异的现象.锦鸡儿属植物对荒漠环境的适应能力和生态恢复作用始终都受到高度的关注[5,31-32].基于以上的研究结论,本研究的主旨设定为:①荒漠豆科锦鸡儿属灌丛下的4个根系微域(根系、根际土、根区土、灌丛间空白土)存在细菌群落多样性和结构组成的层级差异性变化;②探究荒漠豆科锦鸡儿属植物灌丛下根系微域细菌群落结构的层级变异性,及其发生变异的成因. ...
... 该研究于2018年9月在位于毛乌素沙地南缘的宁夏盐池毛乌素沙地生态系统国家定位观测研究站(37°04′—38°10′N,106°30′—107°47′E)开展.研究站位于半旱区,属典型的大陆性季风气候,常年干燥温暖.年平均气温8.1 ℃,日间气温和相对湿度分别是-29.4~37.5 ℃和49%~55%.多年平均降雨量约为292 mm.降雨集中在7—9月,占年降雨量的60%~70%.当地的土壤类型主要为风成沙质土[31].研究区0~1 m剖面土壤质地为砂质,平均容重1.5 g·cm-3.这一地区的生态退化主要是过度放牧、气候变化和风蚀造成的,干旱草原退化为目前的沙地.现在的植被恢复是在2001年通过飞播(油蒿、蒙古岩黄耆和细枝岩黄耆)、植苗扦插种植(小叶锦鸡儿和柠条锦鸡儿)和自然封育建立的[5].目前该区的主要优势旱生灌木种包括油蒿、柠条锦鸡儿、小叶锦鸡儿、蒙古岩黄耆、细枝岩黄耆、沙柳等. ...
1
2014
... 根际土中微生物群落的基因组比植物丰富,也被称为植物的第二基因组[9].根际微生物在调节植物个体的健康、适应性和群落生产力等方面都发挥着难以替代的作用[9,23-26],而植物群落又通过影响土壤养分循环等生态过程影响土壤微生物群落[6].此外,部分根际微生物可以进入植物根系组织形成植物内生菌群落,其群落结构组成与根际土壤微生物群落又有显著的差异性[27-28],这种细菌群落多样性和结构组成在根系组织内和根际土之间的显著差异现象被定义为“根际效应”[29].Sun等[30]发现,毛乌素沙地旱生灌丛下和灌丛间的土壤细菌群落也出现在了细菌群落结构组成显著差异的现象.锦鸡儿属植物对荒漠环境的适应能力和生态恢复作用始终都受到高度的关注[5,31-32].基于以上的研究结论,本研究的主旨设定为:①荒漠豆科锦鸡儿属灌丛下的4个根系微域(根系、根际土、根区土、灌丛间空白土)存在细菌群落多样性和结构组成的层级差异性变化;②探究荒漠豆科锦鸡儿属植物灌丛下根系微域细菌群落结构的层级变异性,及其发生变异的成因. ...
Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota
1
2012
... 小叶锦鸡儿(Caragana microphylla)和柠条锦鸡儿(Caragana korshinskii)在中国西北荒漠地区(包括该研究区内)都是自然共存并广泛分布的优势旱生灌木种.用于采样调查的锦鸡儿属植株均是在2001年通过植苗和扦插种植后自然封育形成的样地内采集的,两种锦鸡儿属植物造林初期植苗混交比例为1∶1.采样时,样地内植株平均株高为1.49 m,平均冠幅1.71 m×1.88 m,行距5 m.灌丛间偶见狗尾草、油蒿、蒺藜等植物.样本是在2018年6月的参试植物花期采集的.参试样本由豆科植物的4个根系微域构成:根系、根际土、根区土、灌丛间空白土.本研究涉及的样本植株主要分布在植苗后自然封育形成的固定沙丘上.选择5个样地(20 m×20 m,间距20 m),在固定的样地内随机选择3株豆科锦鸡儿属灌丛采集植株根系组织和土壤样本,分别将3个植株的样本按照4个根系微域类型划分,各自混合建立一个测试样本.参照Bulgarelli等[33]描述的方法采样.挖至植物根系,用镊子挖取围绕根系1 cm以内的土壤,作为根际土;之后挖出根系并轻轻摇晃植物根部,抖落附属的土壤,带无菌手套将落下的块土(根系1 cm以外的土壤)作为根区土收集在无菌样本袋中;灌丛间空白土则是在灌丛间的裸土下10~40 cm与根区土采样点同一深度采集;根系样本则来自植物根系的二级或三级分支,剪切采集粗细均匀健康无破损的根系5~8 cm于无菌样本袋中. ...
Community structure and diversity of endophytic bacteria in seeds of three consecutive generations of Crotalaria pumila growing on metal mine residues
1
2018
... 依照说明书,使用E.Z.N.A.soil DNA kits (OMEGA,美国),从0.5 g的新鲜样本中提取DNA基因组并于-80 ℃下保存待用.将提取的DNA样本在冰上融化后分别离心并充分混匀,使用Nanodrop超微量分光光度计检测质量,取30 ng进行PCR扩增.使用上游引物F799(5′-AAC_MGG_ATT_AGA_TAC_CCK_G-3′)和下游引物R1193(5′-ACG_TCA_TCC_CCA_CCT_TCC-3′)扩增可变区为V5-V7区的16S rRNA,有研究显示对根系组织测序样本选择扩增可变区V5-V7区,能够有效减少宿主污染[34-36].PCR扩增是在25 μL的反应缓冲液中进行,所述反应缓冲液含有12.5 μL KAPA 2G Robust Hot Start Ready Mix,1 μL Forward Primer(5 μM),1 μL Reverse Primer(5 μM),5 μL DNA(加入的DNA总量为30 ng),最后加入5.5 μL dd H2O补足至25 μL.PCR扩增的流程是:95 ℃下预变性5 min;95 ℃下变性45 s,55 ℃下退火50 s,72 ℃下延伸45 s,28个循环;72 ℃下延伸10 min.PCR产物使用1%琼脂糖凝胶电泳检测扩增目的条带大小,并用Agencourt AMPure XP核酸纯化试剂盒纯化.PCR产物用于构建微生物多样性测序文库,在北京奥维森基因科技有限公司使用Illumina Miseq PE300高通量测序平台进行Paired-end测序. ...
FLASH:fast length adjustment of short reads to improve genome assemblies
1
2011
... 下机数据经过QIIME平台(Version 1.8,http://qiime.org/)根据Barcode序列拆分样本,使用Flash软件(Version 1.2.7)对数据进行过滤、拼接,去除打分低于20、碱基模糊、引物错配序列[35].使用Usearch软件(Version 8.1.1861,http://www.drive5.com/usearch/)去除长度小于230 bp的序列,并根据Gold Database数据库用UCHIME方法[36]比对去除嵌合体序列.最后使用Usearch软件UPARSE算法[37](Version 1.1.579)对优质序列进行OTU聚类(Operational Taxonomic Units-分类操作单元),OTU相似性设置为97%.与Silva128数据库(Release128,http://www.arb-silva.de)使用RDP 分类器算法进行比对,得到每个OTU对应的物种分类信息.Alpha多样性指数的计算同样基于QIIME平台,分析时通常对数据进行抽平处理,以该数据中样本数据最少的数据量作计算,并采用97%的相似度进行分析.各指数的算法如下: ...
UCHIME improves sensitivity and speed of chimera detection
2
2011
... 依照说明书,使用E.Z.N.A.soil DNA kits (OMEGA,美国),从0.5 g的新鲜样本中提取DNA基因组并于-80 ℃下保存待用.将提取的DNA样本在冰上融化后分别离心并充分混匀,使用Nanodrop超微量分光光度计检测质量,取30 ng进行PCR扩增.使用上游引物F799(5′-AAC_MGG_ATT_AGA_TAC_CCK_G-3′)和下游引物R1193(5′-ACG_TCA_TCC_CCA_CCT_TCC-3′)扩增可变区为V5-V7区的16S rRNA,有研究显示对根系组织测序样本选择扩增可变区V5-V7区,能够有效减少宿主污染[34-36].PCR扩增是在25 μL的反应缓冲液中进行,所述反应缓冲液含有12.5 μL KAPA 2G Robust Hot Start Ready Mix,1 μL Forward Primer(5 μM),1 μL Reverse Primer(5 μM),5 μL DNA(加入的DNA总量为30 ng),最后加入5.5 μL dd H2O补足至25 μL.PCR扩增的流程是:95 ℃下预变性5 min;95 ℃下变性45 s,55 ℃下退火50 s,72 ℃下延伸45 s,28个循环;72 ℃下延伸10 min.PCR产物使用1%琼脂糖凝胶电泳检测扩增目的条带大小,并用Agencourt AMPure XP核酸纯化试剂盒纯化.PCR产物用于构建微生物多样性测序文库,在北京奥维森基因科技有限公司使用Illumina Miseq PE300高通量测序平台进行Paired-end测序. ...
... 下机数据经过QIIME平台(Version 1.8,http://qiime.org/)根据Barcode序列拆分样本,使用Flash软件(Version 1.2.7)对数据进行过滤、拼接,去除打分低于20、碱基模糊、引物错配序列[35].使用Usearch软件(Version 8.1.1861,http://www.drive5.com/usearch/)去除长度小于230 bp的序列,并根据Gold Database数据库用UCHIME方法[36]比对去除嵌合体序列.最后使用Usearch软件UPARSE算法[37](Version 1.1.579)对优质序列进行OTU聚类(Operational Taxonomic Units-分类操作单元),OTU相似性设置为97%.与Silva128数据库(Release128,http://www.arb-silva.de)使用RDP 分类器算法进行比对,得到每个OTU对应的物种分类信息.Alpha多样性指数的计算同样基于QIIME平台,分析时通常对数据进行抽平处理,以该数据中样本数据最少的数据量作计算,并采用97%的相似度进行分析.各指数的算法如下: ...
UPARSE:highly accurate OTU sequences from microbial amplicon reads
1
2013
... 下机数据经过QIIME平台(Version 1.8,http://qiime.org/)根据Barcode序列拆分样本,使用Flash软件(Version 1.2.7)对数据进行过滤、拼接,去除打分低于20、碱基模糊、引物错配序列[35].使用Usearch软件(Version 8.1.1861,http://www.drive5.com/usearch/)去除长度小于230 bp的序列,并根据Gold Database数据库用UCHIME方法[36]比对去除嵌合体序列.最后使用Usearch软件UPARSE算法[37](Version 1.1.579)对优质序列进行OTU聚类(Operational Taxonomic Units-分类操作单元),OTU相似性设置为97%.与Silva128数据库(Release128,http://www.arb-silva.de)使用RDP 分类器算法进行比对,得到每个OTU对应的物种分类信息.Alpha多样性指数的计算同样基于QIIME平台,分析时通常对数据进行抽平处理,以该数据中样本数据最少的数据量作计算,并采用97%的相似度进行分析.各指数的算法如下: ...
Rhizobium huakuii sp.nov.isolated from the root nodules of Astragalus sinicus
1
1991
... 本研究采用16S高通量测序技术,分析了荒漠豆科锦鸡儿属植物根系微域中细菌群落在菌物分类学门、目、属水平上的结构和组成,探究了豆科锦鸡儿属植物根系内、根际土、根区土、灌丛间空白土与优势OTUs对应相关的主要贡献细菌群组在门、目、属3个分类学水平上的结构和组成.这极大地扩充了我们对于荒漠豆科锦鸡儿属植物根系微域细菌微生物多样性的认识.这比前人基于特定菌株分离培育鉴定的方法更加全面和精准[38-39].这表明,荒漠豆科锦鸡儿属植物根系内生菌群落结构组成远比我们之前分离培养鉴定的要复杂和多样化. ...
The common nodulation genes of Astragalus sinicus Rhizobia are conserved despite chromosomal diversity
1
2000
... 本研究采用16S高通量测序技术,分析了荒漠豆科锦鸡儿属植物根系微域中细菌群落在菌物分类学门、目、属水平上的结构和组成,探究了豆科锦鸡儿属植物根系内、根际土、根区土、灌丛间空白土与优势OTUs对应相关的主要贡献细菌群组在门、目、属3个分类学水平上的结构和组成.这极大地扩充了我们对于荒漠豆科锦鸡儿属植物根系微域细菌微生物多样性的认识.这比前人基于特定菌株分离培育鉴定的方法更加全面和精准[38-39].这表明,荒漠豆科锦鸡儿属植物根系内生菌群落结构组成远比我们之前分离培养鉴定的要复杂和多样化. ...
Plot-scale manipulations of organic matter inputs to soils correlate with shifts in microbial community composition in a lowland tropical rain forest
1
2010
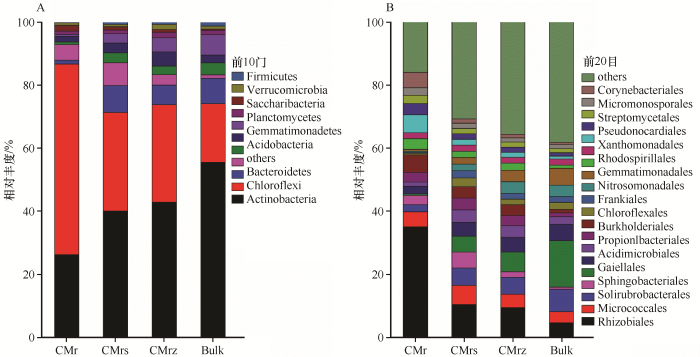
... 本研究检测到在各根系微域中细菌群落的主要贡献细菌群组,在门水平分别为变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、绿弯菌门(Chloroflexi)、拟杆菌门(Bacteroidetes)等(图2).相关研究指出,变形菌门(Proteobacteria)向灌丛下土壤富集的程度与土壤有效碳含量呈正相关,并且其在有效碳含量高的区位与所处生态位异养固氮能效密切相关,即它具有促进土壤中铵态氮被转化利用的作用[40];放线菌门(Actinobacteria)最突出的特性之一是能够产生大量的、种类繁多的抗生素、酶和生长调节剂,在抑制植物疾病、土壤病原体传播等方面发挥着重要作用[41-42],并且其向根系的富集趋势与土壤pH呈正相关[41];绿弯菌门(Chloroflexi)则通过利用硫化物作为电子供体进行光合作用能够导致土壤CO2的减少[43];而灌丛下拟杆菌门(Bacteroidetes)的富集是由于它们能够迅速地利用土壤有机质[44]. ...
Impact of root exudates and plant defense signaling on bacterial communities in the rhizosphere:a review
2
2012
... 本研究检测到在各根系微域中细菌群落的主要贡献细菌群组,在门水平分别为变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、绿弯菌门(Chloroflexi)、拟杆菌门(Bacteroidetes)等(图2).相关研究指出,变形菌门(Proteobacteria)向灌丛下土壤富集的程度与土壤有效碳含量呈正相关,并且其在有效碳含量高的区位与所处生态位异养固氮能效密切相关,即它具有促进土壤中铵态氮被转化利用的作用[40];放线菌门(Actinobacteria)最突出的特性之一是能够产生大量的、种类繁多的抗生素、酶和生长调节剂,在抑制植物疾病、土壤病原体传播等方面发挥着重要作用[41-42],并且其向根系的富集趋势与土壤pH呈正相关[41];绿弯菌门(Chloroflexi)则通过利用硫化物作为电子供体进行光合作用能够导致土壤CO2的减少[43];而灌丛下拟杆菌门(Bacteroidetes)的富集是由于它们能够迅速地利用土壤有机质[44]. ...
... [41];绿弯菌门(Chloroflexi)则通过利用硫化物作为电子供体进行光合作用能够导致土壤CO2的减少[43];而灌丛下拟杆菌门(Bacteroidetes)的富集是由于它们能够迅速地利用土壤有机质[44]. ...
Plant-growth-promoting Rhizobacteria
1
2009
... 本研究检测到在各根系微域中细菌群落的主要贡献细菌群组,在门水平分别为变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、绿弯菌门(Chloroflexi)、拟杆菌门(Bacteroidetes)等(图2).相关研究指出,变形菌门(Proteobacteria)向灌丛下土壤富集的程度与土壤有效碳含量呈正相关,并且其在有效碳含量高的区位与所处生态位异养固氮能效密切相关,即它具有促进土壤中铵态氮被转化利用的作用[40];放线菌门(Actinobacteria)最突出的特性之一是能够产生大量的、种类繁多的抗生素、酶和生长调节剂,在抑制植物疾病、土壤病原体传播等方面发挥着重要作用[41-42],并且其向根系的富集趋势与土壤pH呈正相关[41];绿弯菌门(Chloroflexi)则通过利用硫化物作为电子供体进行光合作用能够导致土壤CO2的减少[43];而灌丛下拟杆菌门(Bacteroidetes)的富集是由于它们能够迅速地利用土壤有机质[44]. ...
Ecological studies of Chloroflexis,a gliding photosynthetic bacterium
1
1973
... 本研究检测到在各根系微域中细菌群落的主要贡献细菌群组,在门水平分别为变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、绿弯菌门(Chloroflexi)、拟杆菌门(Bacteroidetes)等(图2).相关研究指出,变形菌门(Proteobacteria)向灌丛下土壤富集的程度与土壤有效碳含量呈正相关,并且其在有效碳含量高的区位与所处生态位异养固氮能效密切相关,即它具有促进土壤中铵态氮被转化利用的作用[40];放线菌门(Actinobacteria)最突出的特性之一是能够产生大量的、种类繁多的抗生素、酶和生长调节剂,在抑制植物疾病、土壤病原体传播等方面发挥着重要作用[41-42],并且其向根系的富集趋势与土壤pH呈正相关[41];绿弯菌门(Chloroflexi)则通过利用硫化物作为电子供体进行光合作用能够导致土壤CO2的减少[43];而灌丛下拟杆菌门(Bacteroidetes)的富集是由于它们能够迅速地利用土壤有机质[44]. ...
Pyrosequencing analysis for characterization of soil bacterial populations as affected by an integrated livestock-cotton production system
1
2010
... 本研究检测到在各根系微域中细菌群落的主要贡献细菌群组,在门水平分别为变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、绿弯菌门(Chloroflexi)、拟杆菌门(Bacteroidetes)等(图2).相关研究指出,变形菌门(Proteobacteria)向灌丛下土壤富集的程度与土壤有效碳含量呈正相关,并且其在有效碳含量高的区位与所处生态位异养固氮能效密切相关,即它具有促进土壤中铵态氮被转化利用的作用[40];放线菌门(Actinobacteria)最突出的特性之一是能够产生大量的、种类繁多的抗生素、酶和生长调节剂,在抑制植物疾病、土壤病原体传播等方面发挥着重要作用[41-42],并且其向根系的富集趋势与土壤pH呈正相关[41];绿弯菌门(Chloroflexi)则通过利用硫化物作为电子供体进行光合作用能够导致土壤CO2的减少[43];而灌丛下拟杆菌门(Bacteroidetes)的富集是由于它们能够迅速地利用土壤有机质[44]. ...
Soil properties are key determinants for the development of exudate gradients in a rhizosphere simulation model
1
2010
... 荒漠豆科锦鸡儿属植物4个根系微域的样本间Alpha和Beta多样性都存在显著的差异性(P<0.05).荒漠豆科锦鸡儿属植物通过根系微域对灌丛下土壤细菌群落中特定的优势菌群具有筛选富集作用.Beckers等[12]在毛白杨的根系内和根际土生态位的研究中也得到了类似的根系微细域菌群落结构差异性结果,并认为这可能是由于灌丛下的细菌群落是随着根系分布而迁移定殖不均匀造成的.我们的数据表明,植株对根系内定殖的大量内生菌中特定的细菌微生物群组的筛选富集作用是根系内生菌群落发生高度变异性的主要原因.现有的研究表明,向植物根系对特定细菌群组的筛选富集主要通过以下两种途径:①根系分泌物,尤其是植物根际部位分泌含碳代谢产物[45-46];②植物通过化感作用释放化感物质,对土壤细菌微生物产生的化学诱导作用[47].虽然根系分泌物等衍生的营养物质和趋化诱导在根际微域这个生态位广泛存在,但与植物相关的特定有益细菌则需要通过激烈的养分竞争才能成功地迁移并定殖于根系内[48].综上所述,内生菌迁移并定殖于根系内形成一个结构组成相对稳定的群落似乎经历了一个高度变异的过程,这从我们的Alpha多样性(表2)、主成分分析(图1)、根系微域间优势菌群筛选富集分析(图3)、ANOSIM细菌群落结构相似性分析(表3)中都可以得出这样的结果. ...
Soil fungal pathogens and the relationship between plant diversity and productivity
1
2011
... 荒漠豆科锦鸡儿属植物4个根系微域的样本间Alpha和Beta多样性都存在显著的差异性(P<0.05).荒漠豆科锦鸡儿属植物通过根系微域对灌丛下土壤细菌群落中特定的优势菌群具有筛选富集作用.Beckers等[12]在毛白杨的根系内和根际土生态位的研究中也得到了类似的根系微细域菌群落结构差异性结果,并认为这可能是由于灌丛下的细菌群落是随着根系分布而迁移定殖不均匀造成的.我们的数据表明,植株对根系内定殖的大量内生菌中特定的细菌微生物群组的筛选富集作用是根系内生菌群落发生高度变异性的主要原因.现有的研究表明,向植物根系对特定细菌群组的筛选富集主要通过以下两种途径:①根系分泌物,尤其是植物根际部位分泌含碳代谢产物[45-46];②植物通过化感作用释放化感物质,对土壤细菌微生物产生的化学诱导作用[47].虽然根系分泌物等衍生的营养物质和趋化诱导在根际微域这个生态位广泛存在,但与植物相关的特定有益细菌则需要通过激烈的养分竞争才能成功地迁移并定殖于根系内[48].综上所述,内生菌迁移并定殖于根系内形成一个结构组成相对稳定的群落似乎经历了一个高度变异的过程,这从我们的Alpha多样性(表2)、主成分分析(图1)、根系微域间优势菌群筛选富集分析(图3)、ANOSIM细菌群落结构相似性分析(表3)中都可以得出这样的结果. ...
The role of root exudates in rhizosphere interactions with plants and other organisms
1
2006
... 荒漠豆科锦鸡儿属植物4个根系微域的样本间Alpha和Beta多样性都存在显著的差异性(P<0.05).荒漠豆科锦鸡儿属植物通过根系微域对灌丛下土壤细菌群落中特定的优势菌群具有筛选富集作用.Beckers等[12]在毛白杨的根系内和根际土生态位的研究中也得到了类似的根系微细域菌群落结构差异性结果,并认为这可能是由于灌丛下的细菌群落是随着根系分布而迁移定殖不均匀造成的.我们的数据表明,植株对根系内定殖的大量内生菌中特定的细菌微生物群组的筛选富集作用是根系内生菌群落发生高度变异性的主要原因.现有的研究表明,向植物根系对特定细菌群组的筛选富集主要通过以下两种途径:①根系分泌物,尤其是植物根际部位分泌含碳代谢产物[45-46];②植物通过化感作用释放化感物质,对土壤细菌微生物产生的化学诱导作用[47].虽然根系分泌物等衍生的营养物质和趋化诱导在根际微域这个生态位广泛存在,但与植物相关的特定有益细菌则需要通过激烈的养分竞争才能成功地迁移并定殖于根系内[48].综上所述,内生菌迁移并定殖于根系内形成一个结构组成相对稳定的群落似乎经历了一个高度变异的过程,这从我们的Alpha多样性(表2)、主成分分析(图1)、根系微域间优势菌群筛选富集分析(图3)、ANOSIM细菌群落结构相似性分析(表3)中都可以得出这样的结果. ...
Plant growth-promoting bacteria in the rhizo-and endosphere of plants:their role,colonization,mechanisms involved and prospects for utilization
1
2010
... 荒漠豆科锦鸡儿属植物4个根系微域的样本间Alpha和Beta多样性都存在显著的差异性(P<0.05).荒漠豆科锦鸡儿属植物通过根系微域对灌丛下土壤细菌群落中特定的优势菌群具有筛选富集作用.Beckers等[12]在毛白杨的根系内和根际土生态位的研究中也得到了类似的根系微细域菌群落结构差异性结果,并认为这可能是由于灌丛下的细菌群落是随着根系分布而迁移定殖不均匀造成的.我们的数据表明,植株对根系内定殖的大量内生菌中特定的细菌微生物群组的筛选富集作用是根系内生菌群落发生高度变异性的主要原因.现有的研究表明,向植物根系对特定细菌群组的筛选富集主要通过以下两种途径:①根系分泌物,尤其是植物根际部位分泌含碳代谢产物[45-46];②植物通过化感作用释放化感物质,对土壤细菌微生物产生的化学诱导作用[47].虽然根系分泌物等衍生的营养物质和趋化诱导在根际微域这个生态位广泛存在,但与植物相关的特定有益细菌则需要通过激烈的养分竞争才能成功地迁移并定殖于根系内[48].综上所述,内生菌迁移并定殖于根系内形成一个结构组成相对稳定的群落似乎经历了一个高度变异的过程,这从我们的Alpha多样性(表2)、主成分分析(图1)、根系微域间优势菌群筛选富集分析(图3)、ANOSIM细菌群落结构相似性分析(表3)中都可以得出这样的结果. ...
豆科植物和根瘤菌在生态环境中的地位和作用
1
2013
... 我们检测到荒漠豆科锦鸡儿属植物灌丛下根系微域间细菌群落中被逐层筛选富集的根瘤菌属(Rhizobium)和慢生根瘤菌属(Bradyrhizobium)是预期之中的,因为这是已知的能够与豆科植物共生固氮的根瘤菌[49-50].有研究指出,传统认知上豆科植物与根瘤菌的共生关系是具有“宿主专一性”的,即特定的豆科植物只与特定的根瘤菌结瘤固氮.近年来分子生物学基因测序技术表明根瘤菌与豆科植物的共生关系并非传统认知的“宿主专一性”,而是由其共生基因所决定的,根瘤菌凭借共生基因的多样性实际拥有大量的宿主植物,这又被称为“广谱共生”现象[50-51].陈文新[50]提出在应用豆科植物进行植被生态环境恢复时,应考虑到根瘤菌共生基因多样性特点,针对不同的生境和宿主植物制备和选择接种抗逆性强、广谱效益高的根瘤菌菌剂.在被植物筛选富集的其他特定菌属中,贪噬菌属(Variovorax)作为一种植物促生菌既可以促进农作物的生长又可以快速降解土壤中的污染物丙烯酰胺,可以作为一种生物降解剂应用于被丙烯酰胺污染的农田中,对于环境修复具有重要价值[52].鞘脂单胞菌属(Sphingomonas)是一类丰富的新型微生物资源,可用于芳香化合物的生物降解.该属菌株凭借自身的高代谢能力与多功能的生理特性,在环境保护及工业生产方面具有巨大的应用潜力[53]. ...
中国豆科植物根瘤菌资源多样性与系统发育
3
2004
... 我们检测到荒漠豆科锦鸡儿属植物灌丛下根系微域间细菌群落中被逐层筛选富集的根瘤菌属(Rhizobium)和慢生根瘤菌属(Bradyrhizobium)是预期之中的,因为这是已知的能够与豆科植物共生固氮的根瘤菌[49-50].有研究指出,传统认知上豆科植物与根瘤菌的共生关系是具有“宿主专一性”的,即特定的豆科植物只与特定的根瘤菌结瘤固氮.近年来分子生物学基因测序技术表明根瘤菌与豆科植物的共生关系并非传统认知的“宿主专一性”,而是由其共生基因所决定的,根瘤菌凭借共生基因的多样性实际拥有大量的宿主植物,这又被称为“广谱共生”现象[50-51].陈文新[50]提出在应用豆科植物进行植被生态环境恢复时,应考虑到根瘤菌共生基因多样性特点,针对不同的生境和宿主植物制备和选择接种抗逆性强、广谱效益高的根瘤菌菌剂.在被植物筛选富集的其他特定菌属中,贪噬菌属(Variovorax)作为一种植物促生菌既可以促进农作物的生长又可以快速降解土壤中的污染物丙烯酰胺,可以作为一种生物降解剂应用于被丙烯酰胺污染的农田中,对于环境修复具有重要价值[52].鞘脂单胞菌属(Sphingomonas)是一类丰富的新型微生物资源,可用于芳香化合物的生物降解.该属菌株凭借自身的高代谢能力与多功能的生理特性,在环境保护及工业生产方面具有巨大的应用潜力[53]. ...
... [50-51].陈文新[50]提出在应用豆科植物进行植被生态环境恢复时,应考虑到根瘤菌共生基因多样性特点,针对不同的生境和宿主植物制备和选择接种抗逆性强、广谱效益高的根瘤菌菌剂.在被植物筛选富集的其他特定菌属中,贪噬菌属(Variovorax)作为一种植物促生菌既可以促进农作物的生长又可以快速降解土壤中的污染物丙烯酰胺,可以作为一种生物降解剂应用于被丙烯酰胺污染的农田中,对于环境修复具有重要价值[52].鞘脂单胞菌属(Sphingomonas)是一类丰富的新型微生物资源,可用于芳香化合物的生物降解.该属菌株凭借自身的高代谢能力与多功能的生理特性,在环境保护及工业生产方面具有巨大的应用潜力[53]. ...
... [50]提出在应用豆科植物进行植被生态环境恢复时,应考虑到根瘤菌共生基因多样性特点,针对不同的生境和宿主植物制备和选择接种抗逆性强、广谱效益高的根瘤菌菌剂.在被植物筛选富集的其他特定菌属中,贪噬菌属(Variovorax)作为一种植物促生菌既可以促进农作物的生长又可以快速降解土壤中的污染物丙烯酰胺,可以作为一种生物降解剂应用于被丙烯酰胺污染的农田中,对于环境修复具有重要价值[52].鞘脂单胞菌属(Sphingomonas)是一类丰富的新型微生物资源,可用于芳香化合物的生物降解.该属菌株凭借自身的高代谢能力与多功能的生理特性,在环境保护及工业生产方面具有巨大的应用潜力[53]. ...
Molecular basis of symbiotic promiscuity
1
2000
... 我们检测到荒漠豆科锦鸡儿属植物灌丛下根系微域间细菌群落中被逐层筛选富集的根瘤菌属(Rhizobium)和慢生根瘤菌属(Bradyrhizobium)是预期之中的,因为这是已知的能够与豆科植物共生固氮的根瘤菌[49-50].有研究指出,传统认知上豆科植物与根瘤菌的共生关系是具有“宿主专一性”的,即特定的豆科植物只与特定的根瘤菌结瘤固氮.近年来分子生物学基因测序技术表明根瘤菌与豆科植物的共生关系并非传统认知的“宿主专一性”,而是由其共生基因所决定的,根瘤菌凭借共生基因的多样性实际拥有大量的宿主植物,这又被称为“广谱共生”现象[50-51].陈文新[50]提出在应用豆科植物进行植被生态环境恢复时,应考虑到根瘤菌共生基因多样性特点,针对不同的生境和宿主植物制备和选择接种抗逆性强、广谱效益高的根瘤菌菌剂.在被植物筛选富集的其他特定菌属中,贪噬菌属(Variovorax)作为一种植物促生菌既可以促进农作物的生长又可以快速降解土壤中的污染物丙烯酰胺,可以作为一种生物降解剂应用于被丙烯酰胺污染的农田中,对于环境修复具有重要价值[52].鞘脂单胞菌属(Sphingomonas)是一类丰富的新型微生物资源,可用于芳香化合物的生物降解.该属菌株凭借自身的高代谢能力与多功能的生理特性,在环境保护及工业生产方面具有巨大的应用潜力[53]. ...
Acrylamide biodegradation ability and plant growth-promoting properties of Variovoraxboronicumulans CGMCC 4969
1
2013
... 我们检测到荒漠豆科锦鸡儿属植物灌丛下根系微域间细菌群落中被逐层筛选富集的根瘤菌属(Rhizobium)和慢生根瘤菌属(Bradyrhizobium)是预期之中的,因为这是已知的能够与豆科植物共生固氮的根瘤菌[49-50].有研究指出,传统认知上豆科植物与根瘤菌的共生关系是具有“宿主专一性”的,即特定的豆科植物只与特定的根瘤菌结瘤固氮.近年来分子生物学基因测序技术表明根瘤菌与豆科植物的共生关系并非传统认知的“宿主专一性”,而是由其共生基因所决定的,根瘤菌凭借共生基因的多样性实际拥有大量的宿主植物,这又被称为“广谱共生”现象[50-51].陈文新[50]提出在应用豆科植物进行植被生态环境恢复时,应考虑到根瘤菌共生基因多样性特点,针对不同的生境和宿主植物制备和选择接种抗逆性强、广谱效益高的根瘤菌菌剂.在被植物筛选富集的其他特定菌属中,贪噬菌属(Variovorax)作为一种植物促生菌既可以促进农作物的生长又可以快速降解土壤中的污染物丙烯酰胺,可以作为一种生物降解剂应用于被丙烯酰胺污染的农田中,对于环境修复具有重要价值[52].鞘脂单胞菌属(Sphingomonas)是一类丰富的新型微生物资源,可用于芳香化合物的生物降解.该属菌株凭借自身的高代谢能力与多功能的生理特性,在环境保护及工业生产方面具有巨大的应用潜力[53]. ...
The genus Sphingomonas:physiology and ecology
1
1996
... 我们检测到荒漠豆科锦鸡儿属植物灌丛下根系微域间细菌群落中被逐层筛选富集的根瘤菌属(Rhizobium)和慢生根瘤菌属(Bradyrhizobium)是预期之中的,因为这是已知的能够与豆科植物共生固氮的根瘤菌[49-50].有研究指出,传统认知上豆科植物与根瘤菌的共生关系是具有“宿主专一性”的,即特定的豆科植物只与特定的根瘤菌结瘤固氮.近年来分子生物学基因测序技术表明根瘤菌与豆科植物的共生关系并非传统认知的“宿主专一性”,而是由其共生基因所决定的,根瘤菌凭借共生基因的多样性实际拥有大量的宿主植物,这又被称为“广谱共生”现象[50-51].陈文新[50]提出在应用豆科植物进行植被生态环境恢复时,应考虑到根瘤菌共生基因多样性特点,针对不同的生境和宿主植物制备和选择接种抗逆性强、广谱效益高的根瘤菌菌剂.在被植物筛选富集的其他特定菌属中,贪噬菌属(Variovorax)作为一种植物促生菌既可以促进农作物的生长又可以快速降解土壤中的污染物丙烯酰胺,可以作为一种生物降解剂应用于被丙烯酰胺污染的农田中,对于环境修复具有重要价值[52].鞘脂单胞菌属(Sphingomonas)是一类丰富的新型微生物资源,可用于芳香化合物的生物降解.该属菌株凭借自身的高代谢能力与多功能的生理特性,在环境保护及工业生产方面具有巨大的应用潜力[53]. ...





 甘公网安备 62010202000688号
甘公网安备 62010202000688号
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}