浅析中国土地荒漠化生态治理现状、存在问题及对策
1
2017
... 中国的荒漠生态系统面积约2.61×106 km2,占全国国土面积的27.2%,主要分布在水资源匮乏的北方极端干旱、干旱和半干旱地区[1].过去几十年,由于该区域人口的迅速增加和社会、经济发展的需求[2-3],中国北方以耕地面积明显增加为主要标志的绿洲化进程加速[2-5].1990—2015年,西北干旱区耕地面积以年均8.9万hm2的速度持续增加,新增的耕地主要来自对新疆塔里木盆地、伊犁河谷地区和准噶尔盆地荒漠草地的开垦[5].这不仅改变了荒漠原生植被的群落结构,也打破了原有的水热和水盐平衡,导致土壤水分、盐分、养分和物理结构的显著变化[6-10],从而彻底地影响了荒漠生态系统的物质循环和能量流动.以南疆的阿拉尔绿洲为例,这一地区处于极端干旱区,年均降水量不足50 mm,灌溉用水完全依赖天然山脉和昆仑山山脉的冰雪融水,水资源非常匮乏.同时,该地区的土壤盐渍化问题十分严重,农业生产中需要大量灌溉来淋洗土壤中的盐分以满足作物生长.近年来,膜下滴灌技术在作物生长季的推广和应用一定程度上缓解了水资源的匮乏,但每年的冬灌和春灌依然需要大量灌溉用水进行土壤盐分的淋洗[7].过去30年(1990—2019年)耕地面积持续增长,净增长量为729.97 km2,增长率高达312.2%[2].大面积耕地的扩增进一步加剧了塔里木河上游地区本就突出的水土矛盾问题.同时,由于淋洗土壤盐分的灌溉排水导致塔里木河水质的恶化,严重威胁到下游地区的农业生产和生态安全[11-12]. ...
阿拉尔垦区近三十年耕地变化及其驱动因子分析
3
2021
... 中国的荒漠生态系统面积约2.61×106 km2,占全国国土面积的27.2%,主要分布在水资源匮乏的北方极端干旱、干旱和半干旱地区[1].过去几十年,由于该区域人口的迅速增加和社会、经济发展的需求[2-3],中国北方以耕地面积明显增加为主要标志的绿洲化进程加速[2-5].1990—2015年,西北干旱区耕地面积以年均8.9万hm2的速度持续增加,新增的耕地主要来自对新疆塔里木盆地、伊犁河谷地区和准噶尔盆地荒漠草地的开垦[5].这不仅改变了荒漠原生植被的群落结构,也打破了原有的水热和水盐平衡,导致土壤水分、盐分、养分和物理结构的显著变化[6-10],从而彻底地影响了荒漠生态系统的物质循环和能量流动.以南疆的阿拉尔绿洲为例,这一地区处于极端干旱区,年均降水量不足50 mm,灌溉用水完全依赖天然山脉和昆仑山山脉的冰雪融水,水资源非常匮乏.同时,该地区的土壤盐渍化问题十分严重,农业生产中需要大量灌溉来淋洗土壤中的盐分以满足作物生长.近年来,膜下滴灌技术在作物生长季的推广和应用一定程度上缓解了水资源的匮乏,但每年的冬灌和春灌依然需要大量灌溉用水进行土壤盐分的淋洗[7].过去30年(1990—2019年)耕地面积持续增长,净增长量为729.97 km2,增长率高达312.2%[2].大面积耕地的扩增进一步加剧了塔里木河上游地区本就突出的水土矛盾问题.同时,由于淋洗土壤盐分的灌溉排水导致塔里木河水质的恶化,严重威胁到下游地区的农业生产和生态安全[11-12]. ...
... [2-5].1990—2015年,西北干旱区耕地面积以年均8.9万hm2的速度持续增加,新增的耕地主要来自对新疆塔里木盆地、伊犁河谷地区和准噶尔盆地荒漠草地的开垦[5].这不仅改变了荒漠原生植被的群落结构,也打破了原有的水热和水盐平衡,导致土壤水分、盐分、养分和物理结构的显著变化[6-10],从而彻底地影响了荒漠生态系统的物质循环和能量流动.以南疆的阿拉尔绿洲为例,这一地区处于极端干旱区,年均降水量不足50 mm,灌溉用水完全依赖天然山脉和昆仑山山脉的冰雪融水,水资源非常匮乏.同时,该地区的土壤盐渍化问题十分严重,农业生产中需要大量灌溉来淋洗土壤中的盐分以满足作物生长.近年来,膜下滴灌技术在作物生长季的推广和应用一定程度上缓解了水资源的匮乏,但每年的冬灌和春灌依然需要大量灌溉用水进行土壤盐分的淋洗[7].过去30年(1990—2019年)耕地面积持续增长,净增长量为729.97 km2,增长率高达312.2%[2].大面积耕地的扩增进一步加剧了塔里木河上游地区本就突出的水土矛盾问题.同时,由于淋洗土壤盐分的灌溉排水导致塔里木河水质的恶化,严重威胁到下游地区的农业生产和生态安全[11-12]. ...
... [2].大面积耕地的扩增进一步加剧了塔里木河上游地区本就突出的水土矛盾问题.同时,由于淋洗土壤盐分的灌溉排水导致塔里木河水质的恶化,严重威胁到下游地区的农业生产和生态安全[11-12]. ...
中国北方新增耕地的时空变化及驱动因素分区
1
2020
... 中国的荒漠生态系统面积约2.61×106 km2,占全国国土面积的27.2%,主要分布在水资源匮乏的北方极端干旱、干旱和半干旱地区[1].过去几十年,由于该区域人口的迅速增加和社会、经济发展的需求[2-3],中国北方以耕地面积明显增加为主要标志的绿洲化进程加速[2-5].1990—2015年,西北干旱区耕地面积以年均8.9万hm2的速度持续增加,新增的耕地主要来自对新疆塔里木盆地、伊犁河谷地区和准噶尔盆地荒漠草地的开垦[5].这不仅改变了荒漠原生植被的群落结构,也打破了原有的水热和水盐平衡,导致土壤水分、盐分、养分和物理结构的显著变化[6-10],从而彻底地影响了荒漠生态系统的物质循环和能量流动.以南疆的阿拉尔绿洲为例,这一地区处于极端干旱区,年均降水量不足50 mm,灌溉用水完全依赖天然山脉和昆仑山山脉的冰雪融水,水资源非常匮乏.同时,该地区的土壤盐渍化问题十分严重,农业生产中需要大量灌溉来淋洗土壤中的盐分以满足作物生长.近年来,膜下滴灌技术在作物生长季的推广和应用一定程度上缓解了水资源的匮乏,但每年的冬灌和春灌依然需要大量灌溉用水进行土壤盐分的淋洗[7].过去30年(1990—2019年)耕地面积持续增长,净增长量为729.97 km2,增长率高达312.2%[2].大面积耕地的扩增进一步加剧了塔里木河上游地区本就突出的水土矛盾问题.同时,由于淋洗土壤盐分的灌溉排水导致塔里木河水质的恶化,严重威胁到下游地区的农业生产和生态安全[11-12]. ...
2001~2010年中国区域土地利用/覆盖变化对陆面过程影响的模拟研究
2021
基于地貌分区的1990-2015年中国耕地时空特征变化分析
2
2018
... 中国的荒漠生态系统面积约2.61×106 km2,占全国国土面积的27.2%,主要分布在水资源匮乏的北方极端干旱、干旱和半干旱地区[1].过去几十年,由于该区域人口的迅速增加和社会、经济发展的需求[2-3],中国北方以耕地面积明显增加为主要标志的绿洲化进程加速[2-5].1990—2015年,西北干旱区耕地面积以年均8.9万hm2的速度持续增加,新增的耕地主要来自对新疆塔里木盆地、伊犁河谷地区和准噶尔盆地荒漠草地的开垦[5].这不仅改变了荒漠原生植被的群落结构,也打破了原有的水热和水盐平衡,导致土壤水分、盐分、养分和物理结构的显著变化[6-10],从而彻底地影响了荒漠生态系统的物质循环和能量流动.以南疆的阿拉尔绿洲为例,这一地区处于极端干旱区,年均降水量不足50 mm,灌溉用水完全依赖天然山脉和昆仑山山脉的冰雪融水,水资源非常匮乏.同时,该地区的土壤盐渍化问题十分严重,农业生产中需要大量灌溉来淋洗土壤中的盐分以满足作物生长.近年来,膜下滴灌技术在作物生长季的推广和应用一定程度上缓解了水资源的匮乏,但每年的冬灌和春灌依然需要大量灌溉用水进行土壤盐分的淋洗[7].过去30年(1990—2019年)耕地面积持续增长,净增长量为729.97 km2,增长率高达312.2%[2].大面积耕地的扩增进一步加剧了塔里木河上游地区本就突出的水土矛盾问题.同时,由于淋洗土壤盐分的灌溉排水导致塔里木河水质的恶化,严重威胁到下游地区的农业生产和生态安全[11-12]. ...
... [5].这不仅改变了荒漠原生植被的群落结构,也打破了原有的水热和水盐平衡,导致土壤水分、盐分、养分和物理结构的显著变化[6-10],从而彻底地影响了荒漠生态系统的物质循环和能量流动.以南疆的阿拉尔绿洲为例,这一地区处于极端干旱区,年均降水量不足50 mm,灌溉用水完全依赖天然山脉和昆仑山山脉的冰雪融水,水资源非常匮乏.同时,该地区的土壤盐渍化问题十分严重,农业生产中需要大量灌溉来淋洗土壤中的盐分以满足作物生长.近年来,膜下滴灌技术在作物生长季的推广和应用一定程度上缓解了水资源的匮乏,但每年的冬灌和春灌依然需要大量灌溉用水进行土壤盐分的淋洗[7].过去30年(1990—2019年)耕地面积持续增长,净增长量为729.97 km2,增长率高达312.2%[2].大面积耕地的扩增进一步加剧了塔里木河上游地区本就突出的水土矛盾问题.同时,由于淋洗土壤盐分的灌溉排水导致塔里木河水质的恶化,严重威胁到下游地区的农业生产和生态安全[11-12]. ...
干旱区绿洲滴灌对土壤盐碱化的长期影响
1
2014
... 中国的荒漠生态系统面积约2.61×106 km2,占全国国土面积的27.2%,主要分布在水资源匮乏的北方极端干旱、干旱和半干旱地区[1].过去几十年,由于该区域人口的迅速增加和社会、经济发展的需求[2-3],中国北方以耕地面积明显增加为主要标志的绿洲化进程加速[2-5].1990—2015年,西北干旱区耕地面积以年均8.9万hm2的速度持续增加,新增的耕地主要来自对新疆塔里木盆地、伊犁河谷地区和准噶尔盆地荒漠草地的开垦[5].这不仅改变了荒漠原生植被的群落结构,也打破了原有的水热和水盐平衡,导致土壤水分、盐分、养分和物理结构的显著变化[6-10],从而彻底地影响了荒漠生态系统的物质循环和能量流动.以南疆的阿拉尔绿洲为例,这一地区处于极端干旱区,年均降水量不足50 mm,灌溉用水完全依赖天然山脉和昆仑山山脉的冰雪融水,水资源非常匮乏.同时,该地区的土壤盐渍化问题十分严重,农业生产中需要大量灌溉来淋洗土壤中的盐分以满足作物生长.近年来,膜下滴灌技术在作物生长季的推广和应用一定程度上缓解了水资源的匮乏,但每年的冬灌和春灌依然需要大量灌溉用水进行土壤盐分的淋洗[7].过去30年(1990—2019年)耕地面积持续增长,净增长量为729.97 km2,增长率高达312.2%[2].大面积耕地的扩增进一步加剧了塔里木河上游地区本就突出的水土矛盾问题.同时,由于淋洗土壤盐分的灌溉排水导致塔里木河水质的恶化,严重威胁到下游地区的农业生产和生态安全[11-12]. ...
灌溉方式对荒漠绿洲过渡带地下水与土壤理化性质的影响
1
2020
... 中国的荒漠生态系统面积约2.61×106 km2,占全国国土面积的27.2%,主要分布在水资源匮乏的北方极端干旱、干旱和半干旱地区[1].过去几十年,由于该区域人口的迅速增加和社会、经济发展的需求[2-3],中国北方以耕地面积明显增加为主要标志的绿洲化进程加速[2-5].1990—2015年,西北干旱区耕地面积以年均8.9万hm2的速度持续增加,新增的耕地主要来自对新疆塔里木盆地、伊犁河谷地区和准噶尔盆地荒漠草地的开垦[5].这不仅改变了荒漠原生植被的群落结构,也打破了原有的水热和水盐平衡,导致土壤水分、盐分、养分和物理结构的显著变化[6-10],从而彻底地影响了荒漠生态系统的物质循环和能量流动.以南疆的阿拉尔绿洲为例,这一地区处于极端干旱区,年均降水量不足50 mm,灌溉用水完全依赖天然山脉和昆仑山山脉的冰雪融水,水资源非常匮乏.同时,该地区的土壤盐渍化问题十分严重,农业生产中需要大量灌溉来淋洗土壤中的盐分以满足作物生长.近年来,膜下滴灌技术在作物生长季的推广和应用一定程度上缓解了水资源的匮乏,但每年的冬灌和春灌依然需要大量灌溉用水进行土壤盐分的淋洗[7].过去30年(1990—2019年)耕地面积持续增长,净增长量为729.97 km2,增长率高达312.2%[2].大面积耕地的扩增进一步加剧了塔里木河上游地区本就突出的水土矛盾问题.同时,由于淋洗土壤盐分的灌溉排水导致塔里木河水质的恶化,严重威胁到下游地区的农业生产和生态安全[11-12]. ...
开垦对荒漠土壤微生物群落结构特征的影响
9
2017
... 土壤微生物通过参与元素生物地球化学和多种生态过程调节影响植物生长、土壤肥力和健康以及生态系统的结构和功能[13].荒漠土壤微生物群落特征与植被群落(生物量、多样性)和土壤特性(养分水平、盐分、pH值、干旱程度等)密切相关[14-15].由于环境对微生物的选择性差异,处于不同生物气候区和不同植被类型的土壤微生物群落特征存在明显的差异[15-16].阿拉尔绿洲的荒漠因具有极干旱、夏季高温、盐渍化土壤、低覆盖度和多样性盐生灌木分布的生物气候特点,可能孕育了一些耐极端条件(如耐干旱和耐盐碱)的土壤微生物群落,从而塑造了有别于其他荒漠的土壤微生物群落结构.在荒漠开垦为农田后,由于作物种植、灌溉、施肥等农业活动,植被结构、土壤养分、盐分和水分等诸多方面均发生了显著变化[17-18],必然导致土壤微生物群落结构的显著改变[19].学者已从耕作方式[20-21]、施肥制度[21-23]、灌溉方式[24-27]等如何影响典型农区土壤微生物群落方面开展了大量研究,但针对干旱区土地利用方式变化如何影响土壤微生物群落的研究相对较少.少数学者采用培养法[28-29]、变性梯度凝胶电泳法(DGGE)[30]、磷脂脂肪酸法(PLFA)[8]、基因测序[17,31-33]、芯片原位杂交[30]等技术对荒漠及其开垦后土壤细菌或真菌群落生物量、多样性及其结构的变化开展了研究,研究区域主要在盐渍化程度不高的干旱或干旱半干旱区,而针对盐渍化、极干旱荒漠在开垦后土壤微生物群落变化的对比研究较少,有关真菌和古菌群落的研究更为稀缺[34],从而限制了我们对荒漠土壤微生物群落如何响应土地利用方式改变的理解和认识. ...
... 天然荒漠开垦为农田后,作物代替了以盐生型灌木为优势种的原生植被,由于耕作、施肥、灌溉和田间管理等因素,导致农田植被盖度和地上生物量显著提高,而植物丰富度则显著下降.与此同时,开垦后土壤理化属性理应也有明显的变化.通常情况下,干旱荒漠在长期耕作(>10年)后,由于植被残体的输入和肥料的使用,表层土壤TOC和TN等养分显著增加,pH值显著下降[8,17-19],而EC显著下降[19,30]或增加[17].本研究发现,荒漠土壤在连续耕作5年后其EC显著下降,TP和AP显著升高,pH值、TOC和TN及其他C、N组分均没有显著变化(表1).这一结果与上述研究报道不太一致,说明开垦对荒漠土壤属性的影响与其所处的生物气候区、灌溉方式、土壤背景、耕作历史等因素密切相关,短期的耕作对极干旱、盐渍化荒漠植被的影响比土壤属性更为明显,对土壤C、N的累积及其有效性和酸碱度影响不大.可能的原因是,阿拉尔绿洲土壤盐分本底值很高,通常分别在冬季和春季采用大水漫灌方式进行盐分的淋洗,在作物生长季以滴灌方式进行灌溉.5年的耕作历史虽短暂,但由于持续的盐分淋洗后表层土壤盐分含量显著降低,导致土壤EC明显下降.值得注意的是,尽管在棉花生产中有N肥的使用和棉花采收后秸秆还田理论上应该会促进土壤TOC和TN水平提升,但本文却发现开垦5年后土壤中TOC和TN及其组分并无显著变化.北疆荒漠开垦为棉田初期(3—5年)土壤TOC和TN的变化也有类似报道,其TOC含量在开垦后的3—5年内甚至降低[8].其可能的原因如下:①开垦初期由于土壤盐分含量高,棉花的地上生物量较低,通过秸秆转化为土壤TOC和TN的数量相对有限,说明开垦后短期的农业耕作还不足以导致该区域土壤C、N等养分含量的显著提升.②大水漫灌导致土壤中的一些可溶性C、N等养分随排水而流失,也很可能导致C、N等养分沿土壤剖面重新分配,导致表层土壤的TOC和TN与天然荒漠无显著差异.③农事耕作和秸秆还田刺激了土壤呼吸,增加了土壤C的排放[37];而灌溉导致农田土壤水分高于荒漠土壤,有利于反硝化作用而导致N素以N2O等气体的形式释放到大气中[38].但是,磷肥的使用还是导致了土壤中TP和AP成分显著富集,从而增加了P的有效性. ...
... [8].其可能的原因如下:①开垦初期由于土壤盐分含量高,棉花的地上生物量较低,通过秸秆转化为土壤TOC和TN的数量相对有限,说明开垦后短期的农业耕作还不足以导致该区域土壤C、N等养分含量的显著提升.②大水漫灌导致土壤中的一些可溶性C、N等养分随排水而流失,也很可能导致C、N等养分沿土壤剖面重新分配,导致表层土壤的TOC和TN与天然荒漠无显著差异.③农事耕作和秸秆还田刺激了土壤呼吸,增加了土壤C的排放[37];而灌溉导致农田土壤水分高于荒漠土壤,有利于反硝化作用而导致N素以N2O等气体的形式释放到大气中[38].但是,磷肥的使用还是导致了土壤中TP和AP成分显著富集,从而增加了P的有效性. ...
... 一般来讲,土壤水分可利用性和养分水平(如TOC)制约着干旱区土壤微生物群落的生长与繁殖[15,39-40].许多研究发现,土壤细菌和真菌群落生物量与植被盖度和生物量、土壤TOC和TN含量显著正相关[40-41].本研究中,荒漠开垦为农田后其土壤细菌和真菌群落的生物量明显增加,这一研究结果与其他研究报道相一致[8,17,32].主要因为天然荒漠开垦后,绿洲农田植被盖度和生物量显著增加(表1),再加上定期的灌溉,其土壤水热条件得到了极大改善,盐渍化程度大大降低,土壤水分有效性明显增强;同时,农田作物地下根系更为密集,大量的植物残体(秸秆还田)、根系残留物和分泌物输入土壤中,为细菌和真菌群落的生长与繁殖提供了大量的底物.另外,虽然荒漠开垦为农田后,其土壤TN无显著变化,但N和P肥的使用无疑会提高土壤养分的有效性,有利于作物地上和地下生物量的积累,进而促进了细菌和真菌生物量的增加.再者,耕种也增加了表层土壤中的氧气含量,为一些好气型微生物的生理代谢活动创造了更优越的环境.因而,农田土壤细菌和真菌群落生物量明显高于荒漠土壤[8].但是,本研究无法区分耕作、施肥和灌溉对土壤微生物群落特征的各自作用及其机理,有关这方面的研究还需要开展相关的野外控制试验进行深入研究. ...
... [8].但是,本研究无法区分耕作、施肥和灌溉对土壤微生物群落特征的各自作用及其机理,有关这方面的研究还需要开展相关的野外控制试验进行深入研究. ...
... 一些原本适应荒漠环境的细菌和真菌群落丰度在开垦为农田后降低或消失[32],或被更能适应农田环境的微生物群落所替代[33].正如我们的预期,荒漠开垦为农田后土壤细菌群落的多样性显著提升,这一结果与之前的研究报道相一致[8,17,32-33].本文的研究也支持了荒漠土壤细菌和古菌群落多样性随着盐分的增加而降低的研究报道[14],由于高盐分的积累提高了细胞外的渗透压,使许多土壤微生物难以适应渗透压胁迫而被迫死亡或休眠[42-43].然而,开垦对荒漠土壤微生物群落的影响因微生物群落的不同而异,荒漠开垦为农田后将有助于提高土壤细菌和真菌群落的生物量,丰富土壤细菌的群落结构.古菌因其具有嗜热、嗜酸碱和嗜盐的特性,往往能适应荒漠干旱、高温、盐渍化的贫营养性环境[44-45],荒漠开垦为农田后因其适宜生境的改变可能导致一些古菌群落的消失或休眠,从而导致其生物量和Shannon多样性的下降.但是,开垦后土壤真菌群落多样性没有明显变化,这一结果与之前的一些研究报道并不一致[8,17,33].由此可见,盐渍化荒漠土壤细菌群落的生物量和多样性响应土壤利用方式的改变可能比真菌和古菌群落更快、更敏感. ...
... [8,17,33].由此可见,盐渍化荒漠土壤细菌群落的生物量和多样性响应土壤利用方式的改变可能比真菌和古菌群落更快、更敏感. ...
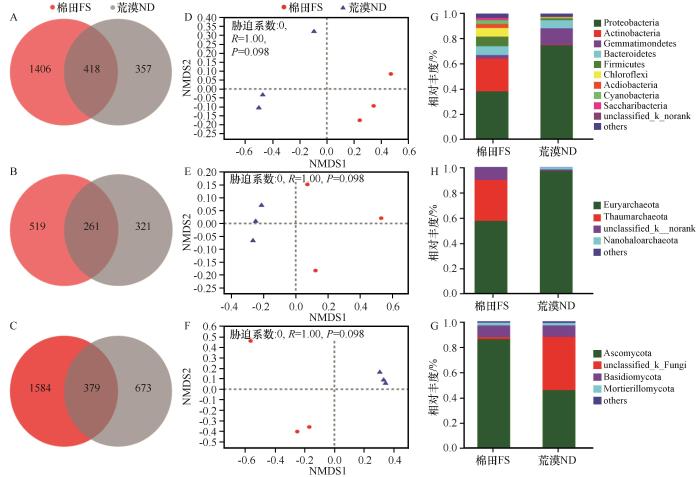
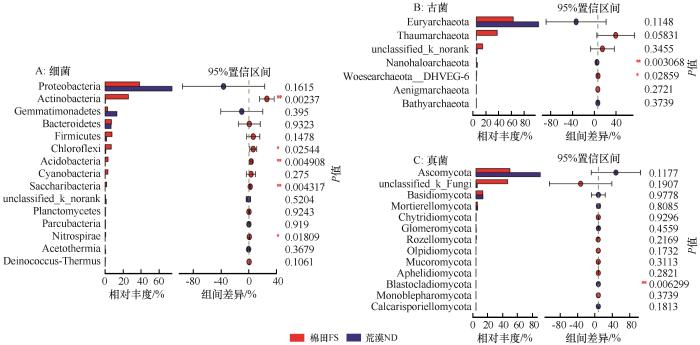
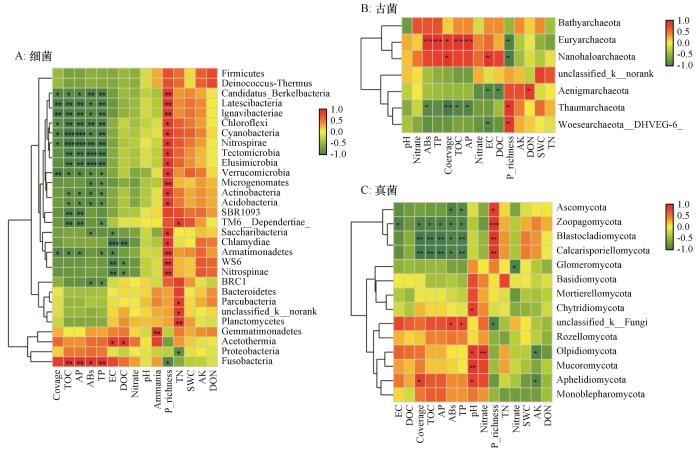
... 本研究中的农田开垦时间仅仅只有短短的5年,但开垦仍然导致土壤微生物群落结构的明显变化,反映出荒漠土壤微生物群落结构对土地利用方式变化的快速响应[8].整体而言,土壤微生物群落结构与植被属性(盖度、生物量和多样性)、土壤EC、TN、TOC、TP和AP显著相关(表3),其强弱关系因微生物类群的不同而异(图5),可能主要由微生物群落的生物学特性所决定.微生物营养假设认为,农业耕作中由于肥料的使用、根系生物量和分泌物等底物的增加,可能会促使一些富营养型微生物相对丰度的增加,而一些贫营养型微生物的丰度降低[30,52].但是,本研究的结果并不支持这一假设.例如,变形菌门和放线菌门往往被认为是富营养型细菌,而酸杆菌门和硝化螺旋菌门(Nitrospirae)则为贫营养型细菌.而此研究中放线菌门、酸杆菌门和硝化螺旋菌门细菌的相对丰度在农田土壤中都显著提高(图3A),这一结果也表明还有其他因素比养分水平在更大程度上决定着因土地利用方式而导致的土壤微生物群落结构的变化,有关这方面的机理还需要进一步的深入研究.同时,最新的一项研究发现,盐分是荒漠土壤细菌和古菌群落结构的关键影响因子,一些耐盐性的细菌和古菌群落存在于盐渍化的荒漠土壤中[14].因此,荒漠在开垦为农田后由于土壤EC的显著减弱,一些耐盐型的微生物,如γ变形菌门(Gammaproteobacteria)细菌和盐杆菌门(Halobacteria)古菌的相对丰度也随之降低.此外,区域尺度[15,53]和控制试验[54]方面的研究发现,水分是影响荒漠土壤微生物群落结构的关键因子.然而,本研究中荒漠和农田土壤的水分没有显著性差异,Mantel test结果也发现土壤水分与微生物群落结构并不相关,表明该区域短期的开垦过程中土壤水分对土壤微生物群落结构的直接作用并不明显,主要是通过影响植被盖度、生物量和土壤EC间接影响土壤微生物的群落构建[15,34].尽管Mantel test分析结果显示土壤TOC和TN仅与细菌群落结构显著正相关,而与古菌和真菌群落结构并不相关(表3),棉田和荒漠土壤之间的TOC和TN也无显著差异(表1),但长期耕作后势必会导致土壤TOC和TN的进一步显著增加[8].众多研究[17,21-23,27]表明长期的施肥和耕作显著改变了土壤微生物群落结构,由此可以预见荒漠农田土壤微生物群落结构可能也势必会进一步发生变化,但由于缺少连续多年的土壤水分、施肥制度和土壤微生物群落变化的相关数据,还无法建立土壤水分动态、施肥制度与土壤微生物群落结构之间的关系.因此,急需通过开展长时间序列的连续动态监测试验进行荒漠土壤微生物群落演替过程及其作用机理方面的研究. ...
... [8].众多研究[17,21-23,27]表明长期的施肥和耕作显著改变了土壤微生物群落结构,由此可以预见荒漠农田土壤微生物群落结构可能也势必会进一步发生变化,但由于缺少连续多年的土壤水分、施肥制度和土壤微生物群落变化的相关数据,还无法建立土壤水分动态、施肥制度与土壤微生物群落结构之间的关系.因此,急需通过开展长时间序列的连续动态监测试验进行荒漠土壤微生物群落演替过程及其作用机理方面的研究. ...
民勤绿洲不同土地利用类型下土壤水盐的空间分布特征分析
2020
新疆阿拉尔灌区土壤次生盐碱化防治及其相关问题研究
1
2007
... 中国的荒漠生态系统面积约2.61×106 km2,占全国国土面积的27.2%,主要分布在水资源匮乏的北方极端干旱、干旱和半干旱地区[1].过去几十年,由于该区域人口的迅速增加和社会、经济发展的需求[2-3],中国北方以耕地面积明显增加为主要标志的绿洲化进程加速[2-5].1990—2015年,西北干旱区耕地面积以年均8.9万hm2的速度持续增加,新增的耕地主要来自对新疆塔里木盆地、伊犁河谷地区和准噶尔盆地荒漠草地的开垦[5].这不仅改变了荒漠原生植被的群落结构,也打破了原有的水热和水盐平衡,导致土壤水分、盐分、养分和物理结构的显著变化[6-10],从而彻底地影响了荒漠生态系统的物质循环和能量流动.以南疆的阿拉尔绿洲为例,这一地区处于极端干旱区,年均降水量不足50 mm,灌溉用水完全依赖天然山脉和昆仑山山脉的冰雪融水,水资源非常匮乏.同时,该地区的土壤盐渍化问题十分严重,农业生产中需要大量灌溉来淋洗土壤中的盐分以满足作物生长.近年来,膜下滴灌技术在作物生长季的推广和应用一定程度上缓解了水资源的匮乏,但每年的冬灌和春灌依然需要大量灌溉用水进行土壤盐分的淋洗[7].过去30年(1990—2019年)耕地面积持续增长,净增长量为729.97 km2,增长率高达312.2%[2].大面积耕地的扩增进一步加剧了塔里木河上游地区本就突出的水土矛盾问题.同时,由于淋洗土壤盐分的灌溉排水导致塔里木河水质的恶化,严重威胁到下游地区的农业生产和生态安全[11-12]. ...
塔里木河下游土地沙漠化及其原因探讨
1
2008
... 中国的荒漠生态系统面积约2.61×106 km2,占全国国土面积的27.2%,主要分布在水资源匮乏的北方极端干旱、干旱和半干旱地区[1].过去几十年,由于该区域人口的迅速增加和社会、经济发展的需求[2-3],中国北方以耕地面积明显增加为主要标志的绿洲化进程加速[2-5].1990—2015年,西北干旱区耕地面积以年均8.9万hm2的速度持续增加,新增的耕地主要来自对新疆塔里木盆地、伊犁河谷地区和准噶尔盆地荒漠草地的开垦[5].这不仅改变了荒漠原生植被的群落结构,也打破了原有的水热和水盐平衡,导致土壤水分、盐分、养分和物理结构的显著变化[6-10],从而彻底地影响了荒漠生态系统的物质循环和能量流动.以南疆的阿拉尔绿洲为例,这一地区处于极端干旱区,年均降水量不足50 mm,灌溉用水完全依赖天然山脉和昆仑山山脉的冰雪融水,水资源非常匮乏.同时,该地区的土壤盐渍化问题十分严重,农业生产中需要大量灌溉来淋洗土壤中的盐分以满足作物生长.近年来,膜下滴灌技术在作物生长季的推广和应用一定程度上缓解了水资源的匮乏,但每年的冬灌和春灌依然需要大量灌溉用水进行土壤盐分的淋洗[7].过去30年(1990—2019年)耕地面积持续增长,净增长量为729.97 km2,增长率高达312.2%[2].大面积耕地的扩增进一步加剧了塔里木河上游地区本就突出的水土矛盾问题.同时,由于淋洗土壤盐分的灌溉排水导致塔里木河水质的恶化,严重威胁到下游地区的农业生产和生态安全[11-12]. ...
塔里木河下游耕地扩张与天然植被退化的定量关系初探
1
2018
... 中国的荒漠生态系统面积约2.61×106 km2,占全国国土面积的27.2%,主要分布在水资源匮乏的北方极端干旱、干旱和半干旱地区[1].过去几十年,由于该区域人口的迅速增加和社会、经济发展的需求[2-3],中国北方以耕地面积明显增加为主要标志的绿洲化进程加速[2-5].1990—2015年,西北干旱区耕地面积以年均8.9万hm2的速度持续增加,新增的耕地主要来自对新疆塔里木盆地、伊犁河谷地区和准噶尔盆地荒漠草地的开垦[5].这不仅改变了荒漠原生植被的群落结构,也打破了原有的水热和水盐平衡,导致土壤水分、盐分、养分和物理结构的显著变化[6-10],从而彻底地影响了荒漠生态系统的物质循环和能量流动.以南疆的阿拉尔绿洲为例,这一地区处于极端干旱区,年均降水量不足50 mm,灌溉用水完全依赖天然山脉和昆仑山山脉的冰雪融水,水资源非常匮乏.同时,该地区的土壤盐渍化问题十分严重,农业生产中需要大量灌溉来淋洗土壤中的盐分以满足作物生长.近年来,膜下滴灌技术在作物生长季的推广和应用一定程度上缓解了水资源的匮乏,但每年的冬灌和春灌依然需要大量灌溉用水进行土壤盐分的淋洗[7].过去30年(1990—2019年)耕地面积持续增长,净增长量为729.97 km2,增长率高达312.2%[2].大面积耕地的扩增进一步加剧了塔里木河上游地区本就突出的水土矛盾问题.同时,由于淋洗土壤盐分的灌溉排水导致塔里木河水质的恶化,严重威胁到下游地区的农业生产和生态安全[11-12]. ...
Microbial indicators for soil quality
1
2018
... 土壤微生物通过参与元素生物地球化学和多种生态过程调节影响植物生长、土壤肥力和健康以及生态系统的结构和功能[13].荒漠土壤微生物群落特征与植被群落(生物量、多样性)和土壤特性(养分水平、盐分、pH值、干旱程度等)密切相关[14-15].由于环境对微生物的选择性差异,处于不同生物气候区和不同植被类型的土壤微生物群落特征存在明显的差异[15-16].阿拉尔绿洲的荒漠因具有极干旱、夏季高温、盐渍化土壤、低覆盖度和多样性盐生灌木分布的生物气候特点,可能孕育了一些耐极端条件(如耐干旱和耐盐碱)的土壤微生物群落,从而塑造了有别于其他荒漠的土壤微生物群落结构.在荒漠开垦为农田后,由于作物种植、灌溉、施肥等农业活动,植被结构、土壤养分、盐分和水分等诸多方面均发生了显著变化[17-18],必然导致土壤微生物群落结构的显著改变[19].学者已从耕作方式[20-21]、施肥制度[21-23]、灌溉方式[24-27]等如何影响典型农区土壤微生物群落方面开展了大量研究,但针对干旱区土地利用方式变化如何影响土壤微生物群落的研究相对较少.少数学者采用培养法[28-29]、变性梯度凝胶电泳法(DGGE)[30]、磷脂脂肪酸法(PLFA)[8]、基因测序[17,31-33]、芯片原位杂交[30]等技术对荒漠及其开垦后土壤细菌或真菌群落生物量、多样性及其结构的变化开展了研究,研究区域主要在盐渍化程度不高的干旱或干旱半干旱区,而针对盐渍化、极干旱荒漠在开垦后土壤微生物群落变化的对比研究较少,有关真菌和古菌群落的研究更为稀缺[34],从而限制了我们对荒漠土壤微生物群落如何响应土地利用方式改变的理解和认识. ...
Salinity Is a key determinant for soil microbial communities in a desert ecosystem
4
2019
... 土壤微生物通过参与元素生物地球化学和多种生态过程调节影响植物生长、土壤肥力和健康以及生态系统的结构和功能[13].荒漠土壤微生物群落特征与植被群落(生物量、多样性)和土壤特性(养分水平、盐分、pH值、干旱程度等)密切相关[14-15].由于环境对微生物的选择性差异,处于不同生物气候区和不同植被类型的土壤微生物群落特征存在明显的差异[15-16].阿拉尔绿洲的荒漠因具有极干旱、夏季高温、盐渍化土壤、低覆盖度和多样性盐生灌木分布的生物气候特点,可能孕育了一些耐极端条件(如耐干旱和耐盐碱)的土壤微生物群落,从而塑造了有别于其他荒漠的土壤微生物群落结构.在荒漠开垦为农田后,由于作物种植、灌溉、施肥等农业活动,植被结构、土壤养分、盐分和水分等诸多方面均发生了显著变化[17-18],必然导致土壤微生物群落结构的显著改变[19].学者已从耕作方式[20-21]、施肥制度[21-23]、灌溉方式[24-27]等如何影响典型农区土壤微生物群落方面开展了大量研究,但针对干旱区土地利用方式变化如何影响土壤微生物群落的研究相对较少.少数学者采用培养法[28-29]、变性梯度凝胶电泳法(DGGE)[30]、磷脂脂肪酸法(PLFA)[8]、基因测序[17,31-33]、芯片原位杂交[30]等技术对荒漠及其开垦后土壤细菌或真菌群落生物量、多样性及其结构的变化开展了研究,研究区域主要在盐渍化程度不高的干旱或干旱半干旱区,而针对盐渍化、极干旱荒漠在开垦后土壤微生物群落变化的对比研究较少,有关真菌和古菌群落的研究更为稀缺[34],从而限制了我们对荒漠土壤微生物群落如何响应土地利用方式改变的理解和认识. ...
... 一些原本适应荒漠环境的细菌和真菌群落丰度在开垦为农田后降低或消失[32],或被更能适应农田环境的微生物群落所替代[33].正如我们的预期,荒漠开垦为农田后土壤细菌群落的多样性显著提升,这一结果与之前的研究报道相一致[8,17,32-33].本文的研究也支持了荒漠土壤细菌和古菌群落多样性随着盐分的增加而降低的研究报道[14],由于高盐分的积累提高了细胞外的渗透压,使许多土壤微生物难以适应渗透压胁迫而被迫死亡或休眠[42-43].然而,开垦对荒漠土壤微生物群落的影响因微生物群落的不同而异,荒漠开垦为农田后将有助于提高土壤细菌和真菌群落的生物量,丰富土壤细菌的群落结构.古菌因其具有嗜热、嗜酸碱和嗜盐的特性,往往能适应荒漠干旱、高温、盐渍化的贫营养性环境[44-45],荒漠开垦为农田后因其适宜生境的改变可能导致一些古菌群落的消失或休眠,从而导致其生物量和Shannon多样性的下降.但是,开垦后土壤真菌群落多样性没有明显变化,这一结果与之前的一些研究报道并不一致[8,17,33].由此可见,盐渍化荒漠土壤细菌群落的生物量和多样性响应土壤利用方式的改变可能比真菌和古菌群落更快、更敏感. ...
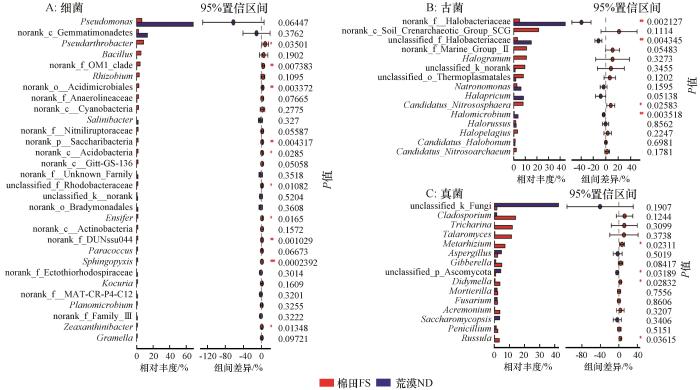
... 本研究发现,荒漠土壤中变形菌门细菌占绝对优势,其相对丰度(74.8%)远高于其他报道(15.0%—40.0%),放线菌门的相对丰度(0.4%)则远低于其他荒漠地区的报道(23%—-52.0%)[14-15,17,31,46-47];厚壁菌门(Firmicutes)细菌也是干旱荒漠中常见的土壤优势细菌,在非洲的Namib沙漠和中国西部戈壁沙漠土壤中的相对丰度高达50%[48]和80%[47],远远高于本研究中的发现(1.5%);芽单孢菌门细菌的相对丰度(13.1%)明显高于腾格里沙漠土壤(<3.0%)[17].与其他研究类似[15,17],子囊菌门真菌是荒漠土壤中的优势真菌,本研究也发现一些其他未分类真菌门在天然荒漠中也占有较高的相对丰度(图2).本研究中荒漠土壤广古菌门古菌的相对丰度(97.0%)远高于北非的Tataouine 沙漠土壤(40.0%)[45].在Atacama 荒漠、Negev沙漠和内蒙古荒漠草原,土壤真菌群落以曲霉菌属(Aspergillus)和青霉菌属(Penicillium)为主[49-51],而本研究中未分类的真菌属在荒漠土壤真菌群落中占绝对优势(图5C).此外,荒漠土壤中还发现了一些未分类的微生物属(图5),有关这些特殊微生物分类方面的研究还需要进一步加强.可见,盐渍化、极干旱的荒漠环境孕育了有别于其他干旱地区的土壤微生物群落结构. ...
... 本研究中的农田开垦时间仅仅只有短短的5年,但开垦仍然导致土壤微生物群落结构的明显变化,反映出荒漠土壤微生物群落结构对土地利用方式变化的快速响应[8].整体而言,土壤微生物群落结构与植被属性(盖度、生物量和多样性)、土壤EC、TN、TOC、TP和AP显著相关(表3),其强弱关系因微生物类群的不同而异(图5),可能主要由微生物群落的生物学特性所决定.微生物营养假设认为,农业耕作中由于肥料的使用、根系生物量和分泌物等底物的增加,可能会促使一些富营养型微生物相对丰度的增加,而一些贫营养型微生物的丰度降低[30,52].但是,本研究的结果并不支持这一假设.例如,变形菌门和放线菌门往往被认为是富营养型细菌,而酸杆菌门和硝化螺旋菌门(Nitrospirae)则为贫营养型细菌.而此研究中放线菌门、酸杆菌门和硝化螺旋菌门细菌的相对丰度在农田土壤中都显著提高(图3A),这一结果也表明还有其他因素比养分水平在更大程度上决定着因土地利用方式而导致的土壤微生物群落结构的变化,有关这方面的机理还需要进一步的深入研究.同时,最新的一项研究发现,盐分是荒漠土壤细菌和古菌群落结构的关键影响因子,一些耐盐性的细菌和古菌群落存在于盐渍化的荒漠土壤中[14].因此,荒漠在开垦为农田后由于土壤EC的显著减弱,一些耐盐型的微生物,如γ变形菌门(Gammaproteobacteria)细菌和盐杆菌门(Halobacteria)古菌的相对丰度也随之降低.此外,区域尺度[15,53]和控制试验[54]方面的研究发现,水分是影响荒漠土壤微生物群落结构的关键因子.然而,本研究中荒漠和农田土壤的水分没有显著性差异,Mantel test结果也发现土壤水分与微生物群落结构并不相关,表明该区域短期的开垦过程中土壤水分对土壤微生物群落结构的直接作用并不明显,主要是通过影响植被盖度、生物量和土壤EC间接影响土壤微生物的群落构建[15,34].尽管Mantel test分析结果显示土壤TOC和TN仅与细菌群落结构显著正相关,而与古菌和真菌群落结构并不相关(表3),棉田和荒漠土壤之间的TOC和TN也无显著差异(表1),但长期耕作后势必会导致土壤TOC和TN的进一步显著增加[8].众多研究[17,21-23,27]表明长期的施肥和耕作显著改变了土壤微生物群落结构,由此可以预见荒漠农田土壤微生物群落结构可能也势必会进一步发生变化,但由于缺少连续多年的土壤水分、施肥制度和土壤微生物群落变化的相关数据,还无法建立土壤水分动态、施肥制度与土壤微生物群落结构之间的关系.因此,急需通过开展长时间序列的连续动态监测试验进行荒漠土壤微生物群落演替过程及其作用机理方面的研究. ...
Increasing aridity reduces soil microbial diversity and abundance in global drylands
7
2015
... 土壤微生物通过参与元素生物地球化学和多种生态过程调节影响植物生长、土壤肥力和健康以及生态系统的结构和功能[13].荒漠土壤微生物群落特征与植被群落(生物量、多样性)和土壤特性(养分水平、盐分、pH值、干旱程度等)密切相关[14-15].由于环境对微生物的选择性差异,处于不同生物气候区和不同植被类型的土壤微生物群落特征存在明显的差异[15-16].阿拉尔绿洲的荒漠因具有极干旱、夏季高温、盐渍化土壤、低覆盖度和多样性盐生灌木分布的生物气候特点,可能孕育了一些耐极端条件(如耐干旱和耐盐碱)的土壤微生物群落,从而塑造了有别于其他荒漠的土壤微生物群落结构.在荒漠开垦为农田后,由于作物种植、灌溉、施肥等农业活动,植被结构、土壤养分、盐分和水分等诸多方面均发生了显著变化[17-18],必然导致土壤微生物群落结构的显著改变[19].学者已从耕作方式[20-21]、施肥制度[21-23]、灌溉方式[24-27]等如何影响典型农区土壤微生物群落方面开展了大量研究,但针对干旱区土地利用方式变化如何影响土壤微生物群落的研究相对较少.少数学者采用培养法[28-29]、变性梯度凝胶电泳法(DGGE)[30]、磷脂脂肪酸法(PLFA)[8]、基因测序[17,31-33]、芯片原位杂交[30]等技术对荒漠及其开垦后土壤细菌或真菌群落生物量、多样性及其结构的变化开展了研究,研究区域主要在盐渍化程度不高的干旱或干旱半干旱区,而针对盐渍化、极干旱荒漠在开垦后土壤微生物群落变化的对比研究较少,有关真菌和古菌群落的研究更为稀缺[34],从而限制了我们对荒漠土壤微生物群落如何响应土地利用方式改变的理解和认识. ...
... [15-16].阿拉尔绿洲的荒漠因具有极干旱、夏季高温、盐渍化土壤、低覆盖度和多样性盐生灌木分布的生物气候特点,可能孕育了一些耐极端条件(如耐干旱和耐盐碱)的土壤微生物群落,从而塑造了有别于其他荒漠的土壤微生物群落结构.在荒漠开垦为农田后,由于作物种植、灌溉、施肥等农业活动,植被结构、土壤养分、盐分和水分等诸多方面均发生了显著变化[17-18],必然导致土壤微生物群落结构的显著改变[19].学者已从耕作方式[20-21]、施肥制度[21-23]、灌溉方式[24-27]等如何影响典型农区土壤微生物群落方面开展了大量研究,但针对干旱区土地利用方式变化如何影响土壤微生物群落的研究相对较少.少数学者采用培养法[28-29]、变性梯度凝胶电泳法(DGGE)[30]、磷脂脂肪酸法(PLFA)[8]、基因测序[17,31-33]、芯片原位杂交[30]等技术对荒漠及其开垦后土壤细菌或真菌群落生物量、多样性及其结构的变化开展了研究,研究区域主要在盐渍化程度不高的干旱或干旱半干旱区,而针对盐渍化、极干旱荒漠在开垦后土壤微生物群落变化的对比研究较少,有关真菌和古菌群落的研究更为稀缺[34],从而限制了我们对荒漠土壤微生物群落如何响应土地利用方式改变的理解和认识. ...
... 一般来讲,土壤水分可利用性和养分水平(如TOC)制约着干旱区土壤微生物群落的生长与繁殖[15,39-40].许多研究发现,土壤细菌和真菌群落生物量与植被盖度和生物量、土壤TOC和TN含量显著正相关[40-41].本研究中,荒漠开垦为农田后其土壤细菌和真菌群落的生物量明显增加,这一研究结果与其他研究报道相一致[8,17,32].主要因为天然荒漠开垦后,绿洲农田植被盖度和生物量显著增加(表1),再加上定期的灌溉,其土壤水热条件得到了极大改善,盐渍化程度大大降低,土壤水分有效性明显增强;同时,农田作物地下根系更为密集,大量的植物残体(秸秆还田)、根系残留物和分泌物输入土壤中,为细菌和真菌群落的生长与繁殖提供了大量的底物.另外,虽然荒漠开垦为农田后,其土壤TN无显著变化,但N和P肥的使用无疑会提高土壤养分的有效性,有利于作物地上和地下生物量的积累,进而促进了细菌和真菌生物量的增加.再者,耕种也增加了表层土壤中的氧气含量,为一些好气型微生物的生理代谢活动创造了更优越的环境.因而,农田土壤细菌和真菌群落生物量明显高于荒漠土壤[8].但是,本研究无法区分耕作、施肥和灌溉对土壤微生物群落特征的各自作用及其机理,有关这方面的研究还需要开展相关的野外控制试验进行深入研究. ...
... 本研究发现,荒漠土壤中变形菌门细菌占绝对优势,其相对丰度(74.8%)远高于其他报道(15.0%—40.0%),放线菌门的相对丰度(0.4%)则远低于其他荒漠地区的报道(23%—-52.0%)[14-15,17,31,46-47];厚壁菌门(Firmicutes)细菌也是干旱荒漠中常见的土壤优势细菌,在非洲的Namib沙漠和中国西部戈壁沙漠土壤中的相对丰度高达50%[48]和80%[47],远远高于本研究中的发现(1.5%);芽单孢菌门细菌的相对丰度(13.1%)明显高于腾格里沙漠土壤(<3.0%)[17].与其他研究类似[15,17],子囊菌门真菌是荒漠土壤中的优势真菌,本研究也发现一些其他未分类真菌门在天然荒漠中也占有较高的相对丰度(图2).本研究中荒漠土壤广古菌门古菌的相对丰度(97.0%)远高于北非的Tataouine 沙漠土壤(40.0%)[45].在Atacama 荒漠、Negev沙漠和内蒙古荒漠草原,土壤真菌群落以曲霉菌属(Aspergillus)和青霉菌属(Penicillium)为主[49-51],而本研究中未分类的真菌属在荒漠土壤真菌群落中占绝对优势(图5C).此外,荒漠土壤中还发现了一些未分类的微生物属(图5),有关这些特殊微生物分类方面的研究还需要进一步加强.可见,盐渍化、极干旱的荒漠环境孕育了有别于其他干旱地区的土壤微生物群落结构. ...
... [15,17],子囊菌门真菌是荒漠土壤中的优势真菌,本研究也发现一些其他未分类真菌门在天然荒漠中也占有较高的相对丰度(图2).本研究中荒漠土壤广古菌门古菌的相对丰度(97.0%)远高于北非的Tataouine 沙漠土壤(40.0%)[45].在Atacama 荒漠、Negev沙漠和内蒙古荒漠草原,土壤真菌群落以曲霉菌属(Aspergillus)和青霉菌属(Penicillium)为主[49-51],而本研究中未分类的真菌属在荒漠土壤真菌群落中占绝对优势(图5C).此外,荒漠土壤中还发现了一些未分类的微生物属(图5),有关这些特殊微生物分类方面的研究还需要进一步加强.可见,盐渍化、极干旱的荒漠环境孕育了有别于其他干旱地区的土壤微生物群落结构. ...
... 本研究中的农田开垦时间仅仅只有短短的5年,但开垦仍然导致土壤微生物群落结构的明显变化,反映出荒漠土壤微生物群落结构对土地利用方式变化的快速响应[8].整体而言,土壤微生物群落结构与植被属性(盖度、生物量和多样性)、土壤EC、TN、TOC、TP和AP显著相关(表3),其强弱关系因微生物类群的不同而异(图5),可能主要由微生物群落的生物学特性所决定.微生物营养假设认为,农业耕作中由于肥料的使用、根系生物量和分泌物等底物的增加,可能会促使一些富营养型微生物相对丰度的增加,而一些贫营养型微生物的丰度降低[30,52].但是,本研究的结果并不支持这一假设.例如,变形菌门和放线菌门往往被认为是富营养型细菌,而酸杆菌门和硝化螺旋菌门(Nitrospirae)则为贫营养型细菌.而此研究中放线菌门、酸杆菌门和硝化螺旋菌门细菌的相对丰度在农田土壤中都显著提高(图3A),这一结果也表明还有其他因素比养分水平在更大程度上决定着因土地利用方式而导致的土壤微生物群落结构的变化,有关这方面的机理还需要进一步的深入研究.同时,最新的一项研究发现,盐分是荒漠土壤细菌和古菌群落结构的关键影响因子,一些耐盐性的细菌和古菌群落存在于盐渍化的荒漠土壤中[14].因此,荒漠在开垦为农田后由于土壤EC的显著减弱,一些耐盐型的微生物,如γ变形菌门(Gammaproteobacteria)细菌和盐杆菌门(Halobacteria)古菌的相对丰度也随之降低.此外,区域尺度[15,53]和控制试验[54]方面的研究发现,水分是影响荒漠土壤微生物群落结构的关键因子.然而,本研究中荒漠和农田土壤的水分没有显著性差异,Mantel test结果也发现土壤水分与微生物群落结构并不相关,表明该区域短期的开垦过程中土壤水分对土壤微生物群落结构的直接作用并不明显,主要是通过影响植被盖度、生物量和土壤EC间接影响土壤微生物的群落构建[15,34].尽管Mantel test分析结果显示土壤TOC和TN仅与细菌群落结构显著正相关,而与古菌和真菌群落结构并不相关(表3),棉田和荒漠土壤之间的TOC和TN也无显著差异(表1),但长期耕作后势必会导致土壤TOC和TN的进一步显著增加[8].众多研究[17,21-23,27]表明长期的施肥和耕作显著改变了土壤微生物群落结构,由此可以预见荒漠农田土壤微生物群落结构可能也势必会进一步发生变化,但由于缺少连续多年的土壤水分、施肥制度和土壤微生物群落变化的相关数据,还无法建立土壤水分动态、施肥制度与土壤微生物群落结构之间的关系.因此,急需通过开展长时间序列的连续动态监测试验进行荒漠土壤微生物群落演替过程及其作用机理方面的研究. ...
... [15,34].尽管Mantel test分析结果显示土壤TOC和TN仅与细菌群落结构显著正相关,而与古菌和真菌群落结构并不相关(表3),棉田和荒漠土壤之间的TOC和TN也无显著差异(表1),但长期耕作后势必会导致土壤TOC和TN的进一步显著增加[8].众多研究[17,21-23,27]表明长期的施肥和耕作显著改变了土壤微生物群落结构,由此可以预见荒漠农田土壤微生物群落结构可能也势必会进一步发生变化,但由于缺少连续多年的土壤水分、施肥制度和土壤微生物群落变化的相关数据,还无法建立土壤水分动态、施肥制度与土壤微生物群落结构之间的关系.因此,急需通过开展长时间序列的连续动态监测试验进行荒漠土壤微生物群落演替过程及其作用机理方面的研究. ...
A global atlas of the dominant bacteria found in soil
1
2018
... 土壤微生物通过参与元素生物地球化学和多种生态过程调节影响植物生长、土壤肥力和健康以及生态系统的结构和功能[13].荒漠土壤微生物群落特征与植被群落(生物量、多样性)和土壤特性(养分水平、盐分、pH值、干旱程度等)密切相关[14-15].由于环境对微生物的选择性差异,处于不同生物气候区和不同植被类型的土壤微生物群落特征存在明显的差异[15-16].阿拉尔绿洲的荒漠因具有极干旱、夏季高温、盐渍化土壤、低覆盖度和多样性盐生灌木分布的生物气候特点,可能孕育了一些耐极端条件(如耐干旱和耐盐碱)的土壤微生物群落,从而塑造了有别于其他荒漠的土壤微生物群落结构.在荒漠开垦为农田后,由于作物种植、灌溉、施肥等农业活动,植被结构、土壤养分、盐分和水分等诸多方面均发生了显著变化[17-18],必然导致土壤微生物群落结构的显著改变[19].学者已从耕作方式[20-21]、施肥制度[21-23]、灌溉方式[24-27]等如何影响典型农区土壤微生物群落方面开展了大量研究,但针对干旱区土地利用方式变化如何影响土壤微生物群落的研究相对较少.少数学者采用培养法[28-29]、变性梯度凝胶电泳法(DGGE)[30]、磷脂脂肪酸法(PLFA)[8]、基因测序[17,31-33]、芯片原位杂交[30]等技术对荒漠及其开垦后土壤细菌或真菌群落生物量、多样性及其结构的变化开展了研究,研究区域主要在盐渍化程度不高的干旱或干旱半干旱区,而针对盐渍化、极干旱荒漠在开垦后土壤微生物群落变化的对比研究较少,有关真菌和古菌群落的研究更为稀缺[34],从而限制了我们对荒漠土壤微生物群落如何响应土地利用方式改变的理解和认识. ...
Change of soil microbial community under long-term fertilization in a reclaimed sandy agricultural ecosystem
11
2019
... 土壤微生物通过参与元素生物地球化学和多种生态过程调节影响植物生长、土壤肥力和健康以及生态系统的结构和功能[13].荒漠土壤微生物群落特征与植被群落(生物量、多样性)和土壤特性(养分水平、盐分、pH值、干旱程度等)密切相关[14-15].由于环境对微生物的选择性差异,处于不同生物气候区和不同植被类型的土壤微生物群落特征存在明显的差异[15-16].阿拉尔绿洲的荒漠因具有极干旱、夏季高温、盐渍化土壤、低覆盖度和多样性盐生灌木分布的生物气候特点,可能孕育了一些耐极端条件(如耐干旱和耐盐碱)的土壤微生物群落,从而塑造了有别于其他荒漠的土壤微生物群落结构.在荒漠开垦为农田后,由于作物种植、灌溉、施肥等农业活动,植被结构、土壤养分、盐分和水分等诸多方面均发生了显著变化[17-18],必然导致土壤微生物群落结构的显著改变[19].学者已从耕作方式[20-21]、施肥制度[21-23]、灌溉方式[24-27]等如何影响典型农区土壤微生物群落方面开展了大量研究,但针对干旱区土地利用方式变化如何影响土壤微生物群落的研究相对较少.少数学者采用培养法[28-29]、变性梯度凝胶电泳法(DGGE)[30]、磷脂脂肪酸法(PLFA)[8]、基因测序[17,31-33]、芯片原位杂交[30]等技术对荒漠及其开垦后土壤细菌或真菌群落生物量、多样性及其结构的变化开展了研究,研究区域主要在盐渍化程度不高的干旱或干旱半干旱区,而针对盐渍化、极干旱荒漠在开垦后土壤微生物群落变化的对比研究较少,有关真菌和古菌群落的研究更为稀缺[34],从而限制了我们对荒漠土壤微生物群落如何响应土地利用方式改变的理解和认识. ...
... [17,31-33]、芯片原位杂交[30]等技术对荒漠及其开垦后土壤细菌或真菌群落生物量、多样性及其结构的变化开展了研究,研究区域主要在盐渍化程度不高的干旱或干旱半干旱区,而针对盐渍化、极干旱荒漠在开垦后土壤微生物群落变化的对比研究较少,有关真菌和古菌群落的研究更为稀缺[34],从而限制了我们对荒漠土壤微生物群落如何响应土地利用方式改变的理解和认识. ...
... 天然荒漠开垦为农田后,作物代替了以盐生型灌木为优势种的原生植被,由于耕作、施肥、灌溉和田间管理等因素,导致农田植被盖度和地上生物量显著提高,而植物丰富度则显著下降.与此同时,开垦后土壤理化属性理应也有明显的变化.通常情况下,干旱荒漠在长期耕作(>10年)后,由于植被残体的输入和肥料的使用,表层土壤TOC和TN等养分显著增加,pH值显著下降[8,17-19],而EC显著下降[19,30]或增加[17].本研究发现,荒漠土壤在连续耕作5年后其EC显著下降,TP和AP显著升高,pH值、TOC和TN及其他C、N组分均没有显著变化(表1).这一结果与上述研究报道不太一致,说明开垦对荒漠土壤属性的影响与其所处的生物气候区、灌溉方式、土壤背景、耕作历史等因素密切相关,短期的耕作对极干旱、盐渍化荒漠植被的影响比土壤属性更为明显,对土壤C、N的累积及其有效性和酸碱度影响不大.可能的原因是,阿拉尔绿洲土壤盐分本底值很高,通常分别在冬季和春季采用大水漫灌方式进行盐分的淋洗,在作物生长季以滴灌方式进行灌溉.5年的耕作历史虽短暂,但由于持续的盐分淋洗后表层土壤盐分含量显著降低,导致土壤EC明显下降.值得注意的是,尽管在棉花生产中有N肥的使用和棉花采收后秸秆还田理论上应该会促进土壤TOC和TN水平提升,但本文却发现开垦5年后土壤中TOC和TN及其组分并无显著变化.北疆荒漠开垦为棉田初期(3—5年)土壤TOC和TN的变化也有类似报道,其TOC含量在开垦后的3—5年内甚至降低[8].其可能的原因如下:①开垦初期由于土壤盐分含量高,棉花的地上生物量较低,通过秸秆转化为土壤TOC和TN的数量相对有限,说明开垦后短期的农业耕作还不足以导致该区域土壤C、N等养分含量的显著提升.②大水漫灌导致土壤中的一些可溶性C、N等养分随排水而流失,也很可能导致C、N等养分沿土壤剖面重新分配,导致表层土壤的TOC和TN与天然荒漠无显著差异.③农事耕作和秸秆还田刺激了土壤呼吸,增加了土壤C的排放[37];而灌溉导致农田土壤水分高于荒漠土壤,有利于反硝化作用而导致N素以N2O等气体的形式释放到大气中[38].但是,磷肥的使用还是导致了土壤中TP和AP成分显著富集,从而增加了P的有效性. ...
... [17].本研究发现,荒漠土壤在连续耕作5年后其EC显著下降,TP和AP显著升高,pH值、TOC和TN及其他C、N组分均没有显著变化(表1).这一结果与上述研究报道不太一致,说明开垦对荒漠土壤属性的影响与其所处的生物气候区、灌溉方式、土壤背景、耕作历史等因素密切相关,短期的耕作对极干旱、盐渍化荒漠植被的影响比土壤属性更为明显,对土壤C、N的累积及其有效性和酸碱度影响不大.可能的原因是,阿拉尔绿洲土壤盐分本底值很高,通常分别在冬季和春季采用大水漫灌方式进行盐分的淋洗,在作物生长季以滴灌方式进行灌溉.5年的耕作历史虽短暂,但由于持续的盐分淋洗后表层土壤盐分含量显著降低,导致土壤EC明显下降.值得注意的是,尽管在棉花生产中有N肥的使用和棉花采收后秸秆还田理论上应该会促进土壤TOC和TN水平提升,但本文却发现开垦5年后土壤中TOC和TN及其组分并无显著变化.北疆荒漠开垦为棉田初期(3—5年)土壤TOC和TN的变化也有类似报道,其TOC含量在开垦后的3—5年内甚至降低[8].其可能的原因如下:①开垦初期由于土壤盐分含量高,棉花的地上生物量较低,通过秸秆转化为土壤TOC和TN的数量相对有限,说明开垦后短期的农业耕作还不足以导致该区域土壤C、N等养分含量的显著提升.②大水漫灌导致土壤中的一些可溶性C、N等养分随排水而流失,也很可能导致C、N等养分沿土壤剖面重新分配,导致表层土壤的TOC和TN与天然荒漠无显著差异.③农事耕作和秸秆还田刺激了土壤呼吸,增加了土壤C的排放[37];而灌溉导致农田土壤水分高于荒漠土壤,有利于反硝化作用而导致N素以N2O等气体的形式释放到大气中[38].但是,磷肥的使用还是导致了土壤中TP和AP成分显著富集,从而增加了P的有效性. ...
... 一般来讲,土壤水分可利用性和养分水平(如TOC)制约着干旱区土壤微生物群落的生长与繁殖[15,39-40].许多研究发现,土壤细菌和真菌群落生物量与植被盖度和生物量、土壤TOC和TN含量显著正相关[40-41].本研究中,荒漠开垦为农田后其土壤细菌和真菌群落的生物量明显增加,这一研究结果与其他研究报道相一致[8,17,32].主要因为天然荒漠开垦后,绿洲农田植被盖度和生物量显著增加(表1),再加上定期的灌溉,其土壤水热条件得到了极大改善,盐渍化程度大大降低,土壤水分有效性明显增强;同时,农田作物地下根系更为密集,大量的植物残体(秸秆还田)、根系残留物和分泌物输入土壤中,为细菌和真菌群落的生长与繁殖提供了大量的底物.另外,虽然荒漠开垦为农田后,其土壤TN无显著变化,但N和P肥的使用无疑会提高土壤养分的有效性,有利于作物地上和地下生物量的积累,进而促进了细菌和真菌生物量的增加.再者,耕种也增加了表层土壤中的氧气含量,为一些好气型微生物的生理代谢活动创造了更优越的环境.因而,农田土壤细菌和真菌群落生物量明显高于荒漠土壤[8].但是,本研究无法区分耕作、施肥和灌溉对土壤微生物群落特征的各自作用及其机理,有关这方面的研究还需要开展相关的野外控制试验进行深入研究. ...
... 一些原本适应荒漠环境的细菌和真菌群落丰度在开垦为农田后降低或消失[32],或被更能适应农田环境的微生物群落所替代[33].正如我们的预期,荒漠开垦为农田后土壤细菌群落的多样性显著提升,这一结果与之前的研究报道相一致[8,17,32-33].本文的研究也支持了荒漠土壤细菌和古菌群落多样性随着盐分的增加而降低的研究报道[14],由于高盐分的积累提高了细胞外的渗透压,使许多土壤微生物难以适应渗透压胁迫而被迫死亡或休眠[42-43].然而,开垦对荒漠土壤微生物群落的影响因微生物群落的不同而异,荒漠开垦为农田后将有助于提高土壤细菌和真菌群落的生物量,丰富土壤细菌的群落结构.古菌因其具有嗜热、嗜酸碱和嗜盐的特性,往往能适应荒漠干旱、高温、盐渍化的贫营养性环境[44-45],荒漠开垦为农田后因其适宜生境的改变可能导致一些古菌群落的消失或休眠,从而导致其生物量和Shannon多样性的下降.但是,开垦后土壤真菌群落多样性没有明显变化,这一结果与之前的一些研究报道并不一致[8,17,33].由此可见,盐渍化荒漠土壤细菌群落的生物量和多样性响应土壤利用方式的改变可能比真菌和古菌群落更快、更敏感. ...
... ,17,33].由此可见,盐渍化荒漠土壤细菌群落的生物量和多样性响应土壤利用方式的改变可能比真菌和古菌群落更快、更敏感. ...
... 本研究发现,荒漠土壤中变形菌门细菌占绝对优势,其相对丰度(74.8%)远高于其他报道(15.0%—40.0%),放线菌门的相对丰度(0.4%)则远低于其他荒漠地区的报道(23%—-52.0%)[14-15,17,31,46-47];厚壁菌门(Firmicutes)细菌也是干旱荒漠中常见的土壤优势细菌,在非洲的Namib沙漠和中国西部戈壁沙漠土壤中的相对丰度高达50%[48]和80%[47],远远高于本研究中的发现(1.5%);芽单孢菌门细菌的相对丰度(13.1%)明显高于腾格里沙漠土壤(<3.0%)[17].与其他研究类似[15,17],子囊菌门真菌是荒漠土壤中的优势真菌,本研究也发现一些其他未分类真菌门在天然荒漠中也占有较高的相对丰度(图2).本研究中荒漠土壤广古菌门古菌的相对丰度(97.0%)远高于北非的Tataouine 沙漠土壤(40.0%)[45].在Atacama 荒漠、Negev沙漠和内蒙古荒漠草原,土壤真菌群落以曲霉菌属(Aspergillus)和青霉菌属(Penicillium)为主[49-51],而本研究中未分类的真菌属在荒漠土壤真菌群落中占绝对优势(图5C).此外,荒漠土壤中还发现了一些未分类的微生物属(图5),有关这些特殊微生物分类方面的研究还需要进一步加强.可见,盐渍化、极干旱的荒漠环境孕育了有别于其他干旱地区的土壤微生物群落结构. ...
... [17].与其他研究类似[15,17],子囊菌门真菌是荒漠土壤中的优势真菌,本研究也发现一些其他未分类真菌门在天然荒漠中也占有较高的相对丰度(图2).本研究中荒漠土壤广古菌门古菌的相对丰度(97.0%)远高于北非的Tataouine 沙漠土壤(40.0%)[45].在Atacama 荒漠、Negev沙漠和内蒙古荒漠草原,土壤真菌群落以曲霉菌属(Aspergillus)和青霉菌属(Penicillium)为主[49-51],而本研究中未分类的真菌属在荒漠土壤真菌群落中占绝对优势(图5C).此外,荒漠土壤中还发现了一些未分类的微生物属(图5),有关这些特殊微生物分类方面的研究还需要进一步加强.可见,盐渍化、极干旱的荒漠环境孕育了有别于其他干旱地区的土壤微生物群落结构. ...
... ,17],子囊菌门真菌是荒漠土壤中的优势真菌,本研究也发现一些其他未分类真菌门在天然荒漠中也占有较高的相对丰度(图2).本研究中荒漠土壤广古菌门古菌的相对丰度(97.0%)远高于北非的Tataouine 沙漠土壤(40.0%)[45].在Atacama 荒漠、Negev沙漠和内蒙古荒漠草原,土壤真菌群落以曲霉菌属(Aspergillus)和青霉菌属(Penicillium)为主[49-51],而本研究中未分类的真菌属在荒漠土壤真菌群落中占绝对优势(图5C).此外,荒漠土壤中还发现了一些未分类的微生物属(图5),有关这些特殊微生物分类方面的研究还需要进一步加强.可见,盐渍化、极干旱的荒漠环境孕育了有别于其他干旱地区的土壤微生物群落结构. ...
... 本研究中的农田开垦时间仅仅只有短短的5年,但开垦仍然导致土壤微生物群落结构的明显变化,反映出荒漠土壤微生物群落结构对土地利用方式变化的快速响应[8].整体而言,土壤微生物群落结构与植被属性(盖度、生物量和多样性)、土壤EC、TN、TOC、TP和AP显著相关(表3),其强弱关系因微生物类群的不同而异(图5),可能主要由微生物群落的生物学特性所决定.微生物营养假设认为,农业耕作中由于肥料的使用、根系生物量和分泌物等底物的增加,可能会促使一些富营养型微生物相对丰度的增加,而一些贫营养型微生物的丰度降低[30,52].但是,本研究的结果并不支持这一假设.例如,变形菌门和放线菌门往往被认为是富营养型细菌,而酸杆菌门和硝化螺旋菌门(Nitrospirae)则为贫营养型细菌.而此研究中放线菌门、酸杆菌门和硝化螺旋菌门细菌的相对丰度在农田土壤中都显著提高(图3A),这一结果也表明还有其他因素比养分水平在更大程度上决定着因土地利用方式而导致的土壤微生物群落结构的变化,有关这方面的机理还需要进一步的深入研究.同时,最新的一项研究发现,盐分是荒漠土壤细菌和古菌群落结构的关键影响因子,一些耐盐性的细菌和古菌群落存在于盐渍化的荒漠土壤中[14].因此,荒漠在开垦为农田后由于土壤EC的显著减弱,一些耐盐型的微生物,如γ变形菌门(Gammaproteobacteria)细菌和盐杆菌门(Halobacteria)古菌的相对丰度也随之降低.此外,区域尺度[15,53]和控制试验[54]方面的研究发现,水分是影响荒漠土壤微生物群落结构的关键因子.然而,本研究中荒漠和农田土壤的水分没有显著性差异,Mantel test结果也发现土壤水分与微生物群落结构并不相关,表明该区域短期的开垦过程中土壤水分对土壤微生物群落结构的直接作用并不明显,主要是通过影响植被盖度、生物量和土壤EC间接影响土壤微生物的群落构建[15,34].尽管Mantel test分析结果显示土壤TOC和TN仅与细菌群落结构显著正相关,而与古菌和真菌群落结构并不相关(表3),棉田和荒漠土壤之间的TOC和TN也无显著差异(表1),但长期耕作后势必会导致土壤TOC和TN的进一步显著增加[8].众多研究[17,21-23,27]表明长期的施肥和耕作显著改变了土壤微生物群落结构,由此可以预见荒漠农田土壤微生物群落结构可能也势必会进一步发生变化,但由于缺少连续多年的土壤水分、施肥制度和土壤微生物群落变化的相关数据,还无法建立土壤水分动态、施肥制度与土壤微生物群落结构之间的关系.因此,急需通过开展长时间序列的连续动态监测试验进行荒漠土壤微生物群落演替过程及其作用机理方面的研究. ...
开垦对西北干旱区荒漠土壤养分含量及主要性质的影响:以甘肃省临泽县为例
1
2008
... 土壤微生物通过参与元素生物地球化学和多种生态过程调节影响植物生长、土壤肥力和健康以及生态系统的结构和功能[13].荒漠土壤微生物群落特征与植被群落(生物量、多样性)和土壤特性(养分水平、盐分、pH值、干旱程度等)密切相关[14-15].由于环境对微生物的选择性差异,处于不同生物气候区和不同植被类型的土壤微生物群落特征存在明显的差异[15-16].阿拉尔绿洲的荒漠因具有极干旱、夏季高温、盐渍化土壤、低覆盖度和多样性盐生灌木分布的生物气候特点,可能孕育了一些耐极端条件(如耐干旱和耐盐碱)的土壤微生物群落,从而塑造了有别于其他荒漠的土壤微生物群落结构.在荒漠开垦为农田后,由于作物种植、灌溉、施肥等农业活动,植被结构、土壤养分、盐分和水分等诸多方面均发生了显著变化[17-18],必然导致土壤微生物群落结构的显著改变[19].学者已从耕作方式[20-21]、施肥制度[21-23]、灌溉方式[24-27]等如何影响典型农区土壤微生物群落方面开展了大量研究,但针对干旱区土地利用方式变化如何影响土壤微生物群落的研究相对较少.少数学者采用培养法[28-29]、变性梯度凝胶电泳法(DGGE)[30]、磷脂脂肪酸法(PLFA)[8]、基因测序[17,31-33]、芯片原位杂交[30]等技术对荒漠及其开垦后土壤细菌或真菌群落生物量、多样性及其结构的变化开展了研究,研究区域主要在盐渍化程度不高的干旱或干旱半干旱区,而针对盐渍化、极干旱荒漠在开垦后土壤微生物群落变化的对比研究较少,有关真菌和古菌群落的研究更为稀缺[34],从而限制了我们对荒漠土壤微生物群落如何响应土地利用方式改变的理解和认识. ...
Profile changes in the soil microbial community when desert becomes oasis
3
2015
... 土壤微生物通过参与元素生物地球化学和多种生态过程调节影响植物生长、土壤肥力和健康以及生态系统的结构和功能[13].荒漠土壤微生物群落特征与植被群落(生物量、多样性)和土壤特性(养分水平、盐分、pH值、干旱程度等)密切相关[14-15].由于环境对微生物的选择性差异,处于不同生物气候区和不同植被类型的土壤微生物群落特征存在明显的差异[15-16].阿拉尔绿洲的荒漠因具有极干旱、夏季高温、盐渍化土壤、低覆盖度和多样性盐生灌木分布的生物气候特点,可能孕育了一些耐极端条件(如耐干旱和耐盐碱)的土壤微生物群落,从而塑造了有别于其他荒漠的土壤微生物群落结构.在荒漠开垦为农田后,由于作物种植、灌溉、施肥等农业活动,植被结构、土壤养分、盐分和水分等诸多方面均发生了显著变化[17-18],必然导致土壤微生物群落结构的显著改变[19].学者已从耕作方式[20-21]、施肥制度[21-23]、灌溉方式[24-27]等如何影响典型农区土壤微生物群落方面开展了大量研究,但针对干旱区土地利用方式变化如何影响土壤微生物群落的研究相对较少.少数学者采用培养法[28-29]、变性梯度凝胶电泳法(DGGE)[30]、磷脂脂肪酸法(PLFA)[8]、基因测序[17,31-33]、芯片原位杂交[30]等技术对荒漠及其开垦后土壤细菌或真菌群落生物量、多样性及其结构的变化开展了研究,研究区域主要在盐渍化程度不高的干旱或干旱半干旱区,而针对盐渍化、极干旱荒漠在开垦后土壤微生物群落变化的对比研究较少,有关真菌和古菌群落的研究更为稀缺[34],从而限制了我们对荒漠土壤微生物群落如何响应土地利用方式改变的理解和认识. ...
... 天然荒漠开垦为农田后,作物代替了以盐生型灌木为优势种的原生植被,由于耕作、施肥、灌溉和田间管理等因素,导致农田植被盖度和地上生物量显著提高,而植物丰富度则显著下降.与此同时,开垦后土壤理化属性理应也有明显的变化.通常情况下,干旱荒漠在长期耕作(>10年)后,由于植被残体的输入和肥料的使用,表层土壤TOC和TN等养分显著增加,pH值显著下降[8,17-19],而EC显著下降[19,30]或增加[17].本研究发现,荒漠土壤在连续耕作5年后其EC显著下降,TP和AP显著升高,pH值、TOC和TN及其他C、N组分均没有显著变化(表1).这一结果与上述研究报道不太一致,说明开垦对荒漠土壤属性的影响与其所处的生物气候区、灌溉方式、土壤背景、耕作历史等因素密切相关,短期的耕作对极干旱、盐渍化荒漠植被的影响比土壤属性更为明显,对土壤C、N的累积及其有效性和酸碱度影响不大.可能的原因是,阿拉尔绿洲土壤盐分本底值很高,通常分别在冬季和春季采用大水漫灌方式进行盐分的淋洗,在作物生长季以滴灌方式进行灌溉.5年的耕作历史虽短暂,但由于持续的盐分淋洗后表层土壤盐分含量显著降低,导致土壤EC明显下降.值得注意的是,尽管在棉花生产中有N肥的使用和棉花采收后秸秆还田理论上应该会促进土壤TOC和TN水平提升,但本文却发现开垦5年后土壤中TOC和TN及其组分并无显著变化.北疆荒漠开垦为棉田初期(3—5年)土壤TOC和TN的变化也有类似报道,其TOC含量在开垦后的3—5年内甚至降低[8].其可能的原因如下:①开垦初期由于土壤盐分含量高,棉花的地上生物量较低,通过秸秆转化为土壤TOC和TN的数量相对有限,说明开垦后短期的农业耕作还不足以导致该区域土壤C、N等养分含量的显著提升.②大水漫灌导致土壤中的一些可溶性C、N等养分随排水而流失,也很可能导致C、N等养分沿土壤剖面重新分配,导致表层土壤的TOC和TN与天然荒漠无显著差异.③农事耕作和秸秆还田刺激了土壤呼吸,增加了土壤C的排放[37];而灌溉导致农田土壤水分高于荒漠土壤,有利于反硝化作用而导致N素以N2O等气体的形式释放到大气中[38].但是,磷肥的使用还是导致了土壤中TP和AP成分显著富集,从而增加了P的有效性. ...
... [19,30]或增加[17].本研究发现,荒漠土壤在连续耕作5年后其EC显著下降,TP和AP显著升高,pH值、TOC和TN及其他C、N组分均没有显著变化(表1).这一结果与上述研究报道不太一致,说明开垦对荒漠土壤属性的影响与其所处的生物气候区、灌溉方式、土壤背景、耕作历史等因素密切相关,短期的耕作对极干旱、盐渍化荒漠植被的影响比土壤属性更为明显,对土壤C、N的累积及其有效性和酸碱度影响不大.可能的原因是,阿拉尔绿洲土壤盐分本底值很高,通常分别在冬季和春季采用大水漫灌方式进行盐分的淋洗,在作物生长季以滴灌方式进行灌溉.5年的耕作历史虽短暂,但由于持续的盐分淋洗后表层土壤盐分含量显著降低,导致土壤EC明显下降.值得注意的是,尽管在棉花生产中有N肥的使用和棉花采收后秸秆还田理论上应该会促进土壤TOC和TN水平提升,但本文却发现开垦5年后土壤中TOC和TN及其组分并无显著变化.北疆荒漠开垦为棉田初期(3—5年)土壤TOC和TN的变化也有类似报道,其TOC含量在开垦后的3—5年内甚至降低[8].其可能的原因如下:①开垦初期由于土壤盐分含量高,棉花的地上生物量较低,通过秸秆转化为土壤TOC和TN的数量相对有限,说明开垦后短期的农业耕作还不足以导致该区域土壤C、N等养分含量的显著提升.②大水漫灌导致土壤中的一些可溶性C、N等养分随排水而流失,也很可能导致C、N等养分沿土壤剖面重新分配,导致表层土壤的TOC和TN与天然荒漠无显著差异.③农事耕作和秸秆还田刺激了土壤呼吸,增加了土壤C的排放[37];而灌溉导致农田土壤水分高于荒漠土壤,有利于反硝化作用而导致N素以N2O等气体的形式释放到大气中[38].但是,磷肥的使用还是导致了土壤中TP和AP成分显著富集,从而增加了P的有效性. ...
Changes in soil microbial properties with no-tillage in Chinese cropping systems
1
2013
... 土壤微生物通过参与元素生物地球化学和多种生态过程调节影响植物生长、土壤肥力和健康以及生态系统的结构和功能[13].荒漠土壤微生物群落特征与植被群落(生物量、多样性)和土壤特性(养分水平、盐分、pH值、干旱程度等)密切相关[14-15].由于环境对微生物的选择性差异,处于不同生物气候区和不同植被类型的土壤微生物群落特征存在明显的差异[15-16].阿拉尔绿洲的荒漠因具有极干旱、夏季高温、盐渍化土壤、低覆盖度和多样性盐生灌木分布的生物气候特点,可能孕育了一些耐极端条件(如耐干旱和耐盐碱)的土壤微生物群落,从而塑造了有别于其他荒漠的土壤微生物群落结构.在荒漠开垦为农田后,由于作物种植、灌溉、施肥等农业活动,植被结构、土壤养分、盐分和水分等诸多方面均发生了显著变化[17-18],必然导致土壤微生物群落结构的显著改变[19].学者已从耕作方式[20-21]、施肥制度[21-23]、灌溉方式[24-27]等如何影响典型农区土壤微生物群落方面开展了大量研究,但针对干旱区土地利用方式变化如何影响土壤微生物群落的研究相对较少.少数学者采用培养法[28-29]、变性梯度凝胶电泳法(DGGE)[30]、磷脂脂肪酸法(PLFA)[8]、基因测序[17,31-33]、芯片原位杂交[30]等技术对荒漠及其开垦后土壤细菌或真菌群落生物量、多样性及其结构的变化开展了研究,研究区域主要在盐渍化程度不高的干旱或干旱半干旱区,而针对盐渍化、极干旱荒漠在开垦后土壤微生物群落变化的对比研究较少,有关真菌和古菌群落的研究更为稀缺[34],从而限制了我们对荒漠土壤微生物群落如何响应土地利用方式改变的理解和认识. ...
Soil microbial diversity under different fertilization and tillage practices:a review
3
2010
... 土壤微生物通过参与元素生物地球化学和多种生态过程调节影响植物生长、土壤肥力和健康以及生态系统的结构和功能[13].荒漠土壤微生物群落特征与植被群落(生物量、多样性)和土壤特性(养分水平、盐分、pH值、干旱程度等)密切相关[14-15].由于环境对微生物的选择性差异,处于不同生物气候区和不同植被类型的土壤微生物群落特征存在明显的差异[15-16].阿拉尔绿洲的荒漠因具有极干旱、夏季高温、盐渍化土壤、低覆盖度和多样性盐生灌木分布的生物气候特点,可能孕育了一些耐极端条件(如耐干旱和耐盐碱)的土壤微生物群落,从而塑造了有别于其他荒漠的土壤微生物群落结构.在荒漠开垦为农田后,由于作物种植、灌溉、施肥等农业活动,植被结构、土壤养分、盐分和水分等诸多方面均发生了显著变化[17-18],必然导致土壤微生物群落结构的显著改变[19].学者已从耕作方式[20-21]、施肥制度[21-23]、灌溉方式[24-27]等如何影响典型农区土壤微生物群落方面开展了大量研究,但针对干旱区土地利用方式变化如何影响土壤微生物群落的研究相对较少.少数学者采用培养法[28-29]、变性梯度凝胶电泳法(DGGE)[30]、磷脂脂肪酸法(PLFA)[8]、基因测序[17,31-33]、芯片原位杂交[30]等技术对荒漠及其开垦后土壤细菌或真菌群落生物量、多样性及其结构的变化开展了研究,研究区域主要在盐渍化程度不高的干旱或干旱半干旱区,而针对盐渍化、极干旱荒漠在开垦后土壤微生物群落变化的对比研究较少,有关真菌和古菌群落的研究更为稀缺[34],从而限制了我们对荒漠土壤微生物群落如何响应土地利用方式改变的理解和认识. ...
... [21-23]、灌溉方式[24-27]等如何影响典型农区土壤微生物群落方面开展了大量研究,但针对干旱区土地利用方式变化如何影响土壤微生物群落的研究相对较少.少数学者采用培养法[28-29]、变性梯度凝胶电泳法(DGGE)[30]、磷脂脂肪酸法(PLFA)[8]、基因测序[17,31-33]、芯片原位杂交[30]等技术对荒漠及其开垦后土壤细菌或真菌群落生物量、多样性及其结构的变化开展了研究,研究区域主要在盐渍化程度不高的干旱或干旱半干旱区,而针对盐渍化、极干旱荒漠在开垦后土壤微生物群落变化的对比研究较少,有关真菌和古菌群落的研究更为稀缺[34],从而限制了我们对荒漠土壤微生物群落如何响应土地利用方式改变的理解和认识. ...
... 本研究中的农田开垦时间仅仅只有短短的5年,但开垦仍然导致土壤微生物群落结构的明显变化,反映出荒漠土壤微生物群落结构对土地利用方式变化的快速响应[8].整体而言,土壤微生物群落结构与植被属性(盖度、生物量和多样性)、土壤EC、TN、TOC、TP和AP显著相关(表3),其强弱关系因微生物类群的不同而异(图5),可能主要由微生物群落的生物学特性所决定.微生物营养假设认为,农业耕作中由于肥料的使用、根系生物量和分泌物等底物的增加,可能会促使一些富营养型微生物相对丰度的增加,而一些贫营养型微生物的丰度降低[30,52].但是,本研究的结果并不支持这一假设.例如,变形菌门和放线菌门往往被认为是富营养型细菌,而酸杆菌门和硝化螺旋菌门(Nitrospirae)则为贫营养型细菌.而此研究中放线菌门、酸杆菌门和硝化螺旋菌门细菌的相对丰度在农田土壤中都显著提高(图3A),这一结果也表明还有其他因素比养分水平在更大程度上决定着因土地利用方式而导致的土壤微生物群落结构的变化,有关这方面的机理还需要进一步的深入研究.同时,最新的一项研究发现,盐分是荒漠土壤细菌和古菌群落结构的关键影响因子,一些耐盐性的细菌和古菌群落存在于盐渍化的荒漠土壤中[14].因此,荒漠在开垦为农田后由于土壤EC的显著减弱,一些耐盐型的微生物,如γ变形菌门(Gammaproteobacteria)细菌和盐杆菌门(Halobacteria)古菌的相对丰度也随之降低.此外,区域尺度[15,53]和控制试验[54]方面的研究发现,水分是影响荒漠土壤微生物群落结构的关键因子.然而,本研究中荒漠和农田土壤的水分没有显著性差异,Mantel test结果也发现土壤水分与微生物群落结构并不相关,表明该区域短期的开垦过程中土壤水分对土壤微生物群落结构的直接作用并不明显,主要是通过影响植被盖度、生物量和土壤EC间接影响土壤微生物的群落构建[15,34].尽管Mantel test分析结果显示土壤TOC和TN仅与细菌群落结构显著正相关,而与古菌和真菌群落结构并不相关(表3),棉田和荒漠土壤之间的TOC和TN也无显著差异(表1),但长期耕作后势必会导致土壤TOC和TN的进一步显著增加[8].众多研究[17,21-23,27]表明长期的施肥和耕作显著改变了土壤微生物群落结构,由此可以预见荒漠农田土壤微生物群落结构可能也势必会进一步发生变化,但由于缺少连续多年的土壤水分、施肥制度和土壤微生物群落变化的相关数据,还无法建立土壤水分动态、施肥制度与土壤微生物群落结构之间的关系.因此,急需通过开展长时间序列的连续动态监测试验进行荒漠土壤微生物群落演替过程及其作用机理方面的研究. ...
Effects of long-term fertilization management practices on soil microbial biomass in China's cropland:a meta-analysis
2017
Assessment of the impact of different fertilization systems on soil microbial ecology
2
2010
... 土壤微生物通过参与元素生物地球化学和多种生态过程调节影响植物生长、土壤肥力和健康以及生态系统的结构和功能[13].荒漠土壤微生物群落特征与植被群落(生物量、多样性)和土壤特性(养分水平、盐分、pH值、干旱程度等)密切相关[14-15].由于环境对微生物的选择性差异,处于不同生物气候区和不同植被类型的土壤微生物群落特征存在明显的差异[15-16].阿拉尔绿洲的荒漠因具有极干旱、夏季高温、盐渍化土壤、低覆盖度和多样性盐生灌木分布的生物气候特点,可能孕育了一些耐极端条件(如耐干旱和耐盐碱)的土壤微生物群落,从而塑造了有别于其他荒漠的土壤微生物群落结构.在荒漠开垦为农田后,由于作物种植、灌溉、施肥等农业活动,植被结构、土壤养分、盐分和水分等诸多方面均发生了显著变化[17-18],必然导致土壤微生物群落结构的显著改变[19].学者已从耕作方式[20-21]、施肥制度[21-23]、灌溉方式[24-27]等如何影响典型农区土壤微生物群落方面开展了大量研究,但针对干旱区土地利用方式变化如何影响土壤微生物群落的研究相对较少.少数学者采用培养法[28-29]、变性梯度凝胶电泳法(DGGE)[30]、磷脂脂肪酸法(PLFA)[8]、基因测序[17,31-33]、芯片原位杂交[30]等技术对荒漠及其开垦后土壤细菌或真菌群落生物量、多样性及其结构的变化开展了研究,研究区域主要在盐渍化程度不高的干旱或干旱半干旱区,而针对盐渍化、极干旱荒漠在开垦后土壤微生物群落变化的对比研究较少,有关真菌和古菌群落的研究更为稀缺[34],从而限制了我们对荒漠土壤微生物群落如何响应土地利用方式改变的理解和认识. ...
... 本研究中的农田开垦时间仅仅只有短短的5年,但开垦仍然导致土壤微生物群落结构的明显变化,反映出荒漠土壤微生物群落结构对土地利用方式变化的快速响应[8].整体而言,土壤微生物群落结构与植被属性(盖度、生物量和多样性)、土壤EC、TN、TOC、TP和AP显著相关(表3),其强弱关系因微生物类群的不同而异(图5),可能主要由微生物群落的生物学特性所决定.微生物营养假设认为,农业耕作中由于肥料的使用、根系生物量和分泌物等底物的增加,可能会促使一些富营养型微生物相对丰度的增加,而一些贫营养型微生物的丰度降低[30,52].但是,本研究的结果并不支持这一假设.例如,变形菌门和放线菌门往往被认为是富营养型细菌,而酸杆菌门和硝化螺旋菌门(Nitrospirae)则为贫营养型细菌.而此研究中放线菌门、酸杆菌门和硝化螺旋菌门细菌的相对丰度在农田土壤中都显著提高(图3A),这一结果也表明还有其他因素比养分水平在更大程度上决定着因土地利用方式而导致的土壤微生物群落结构的变化,有关这方面的机理还需要进一步的深入研究.同时,最新的一项研究发现,盐分是荒漠土壤细菌和古菌群落结构的关键影响因子,一些耐盐性的细菌和古菌群落存在于盐渍化的荒漠土壤中[14].因此,荒漠在开垦为农田后由于土壤EC的显著减弱,一些耐盐型的微生物,如γ变形菌门(Gammaproteobacteria)细菌和盐杆菌门(Halobacteria)古菌的相对丰度也随之降低.此外,区域尺度[15,53]和控制试验[54]方面的研究发现,水分是影响荒漠土壤微生物群落结构的关键因子.然而,本研究中荒漠和农田土壤的水分没有显著性差异,Mantel test结果也发现土壤水分与微生物群落结构并不相关,表明该区域短期的开垦过程中土壤水分对土壤微生物群落结构的直接作用并不明显,主要是通过影响植被盖度、生物量和土壤EC间接影响土壤微生物的群落构建[15,34].尽管Mantel test分析结果显示土壤TOC和TN仅与细菌群落结构显著正相关,而与古菌和真菌群落结构并不相关(表3),棉田和荒漠土壤之间的TOC和TN也无显著差异(表1),但长期耕作后势必会导致土壤TOC和TN的进一步显著增加[8].众多研究[17,21-23,27]表明长期的施肥和耕作显著改变了土壤微生物群落结构,由此可以预见荒漠农田土壤微生物群落结构可能也势必会进一步发生变化,但由于缺少连续多年的土壤水分、施肥制度和土壤微生物群落变化的相关数据,还无法建立土壤水分动态、施肥制度与土壤微生物群落结构之间的关系.因此,急需通过开展长时间序列的连续动态监测试验进行荒漠土壤微生物群落演替过程及其作用机理方面的研究. ...
Effects of long-term sewage irrigation on agricultural soil microbial structural and functional characterizations in Shandong,China
1
2008
... 土壤微生物通过参与元素生物地球化学和多种生态过程调节影响植物生长、土壤肥力和健康以及生态系统的结构和功能[13].荒漠土壤微生物群落特征与植被群落(生物量、多样性)和土壤特性(养分水平、盐分、pH值、干旱程度等)密切相关[14-15].由于环境对微生物的选择性差异,处于不同生物气候区和不同植被类型的土壤微生物群落特征存在明显的差异[15-16].阿拉尔绿洲的荒漠因具有极干旱、夏季高温、盐渍化土壤、低覆盖度和多样性盐生灌木分布的生物气候特点,可能孕育了一些耐极端条件(如耐干旱和耐盐碱)的土壤微生物群落,从而塑造了有别于其他荒漠的土壤微生物群落结构.在荒漠开垦为农田后,由于作物种植、灌溉、施肥等农业活动,植被结构、土壤养分、盐分和水分等诸多方面均发生了显著变化[17-18],必然导致土壤微生物群落结构的显著改变[19].学者已从耕作方式[20-21]、施肥制度[21-23]、灌溉方式[24-27]等如何影响典型农区土壤微生物群落方面开展了大量研究,但针对干旱区土地利用方式变化如何影响土壤微生物群落的研究相对较少.少数学者采用培养法[28-29]、变性梯度凝胶电泳法(DGGE)[30]、磷脂脂肪酸法(PLFA)[8]、基因测序[17,31-33]、芯片原位杂交[30]等技术对荒漠及其开垦后土壤细菌或真菌群落生物量、多样性及其结构的变化开展了研究,研究区域主要在盐渍化程度不高的干旱或干旱半干旱区,而针对盐渍化、极干旱荒漠在开垦后土壤微生物群落变化的对比研究较少,有关真菌和古菌群落的研究更为稀缺[34],从而限制了我们对荒漠土壤微生物群落如何响应土地利用方式改变的理解和认识. ...
Combined effects of reduced irrigation and water quality on the soil microbial community of a citrus orchard under semi-arid conditions
2017
Impact of treated wastewater for irrigation on soil microbial communities
2018
Effects of irrigation patterns and nitrogen fertilization on rice yield and microbial community structure in paddy soil
2
2012
... 土壤微生物通过参与元素生物地球化学和多种生态过程调节影响植物生长、土壤肥力和健康以及生态系统的结构和功能[13].荒漠土壤微生物群落特征与植被群落(生物量、多样性)和土壤特性(养分水平、盐分、pH值、干旱程度等)密切相关[14-15].由于环境对微生物的选择性差异,处于不同生物气候区和不同植被类型的土壤微生物群落特征存在明显的差异[15-16].阿拉尔绿洲的荒漠因具有极干旱、夏季高温、盐渍化土壤、低覆盖度和多样性盐生灌木分布的生物气候特点,可能孕育了一些耐极端条件(如耐干旱和耐盐碱)的土壤微生物群落,从而塑造了有别于其他荒漠的土壤微生物群落结构.在荒漠开垦为农田后,由于作物种植、灌溉、施肥等农业活动,植被结构、土壤养分、盐分和水分等诸多方面均发生了显著变化[17-18],必然导致土壤微生物群落结构的显著改变[19].学者已从耕作方式[20-21]、施肥制度[21-23]、灌溉方式[24-27]等如何影响典型农区土壤微生物群落方面开展了大量研究,但针对干旱区土地利用方式变化如何影响土壤微生物群落的研究相对较少.少数学者采用培养法[28-29]、变性梯度凝胶电泳法(DGGE)[30]、磷脂脂肪酸法(PLFA)[8]、基因测序[17,31-33]、芯片原位杂交[30]等技术对荒漠及其开垦后土壤细菌或真菌群落生物量、多样性及其结构的变化开展了研究,研究区域主要在盐渍化程度不高的干旱或干旱半干旱区,而针对盐渍化、极干旱荒漠在开垦后土壤微生物群落变化的对比研究较少,有关真菌和古菌群落的研究更为稀缺[34],从而限制了我们对荒漠土壤微生物群落如何响应土地利用方式改变的理解和认识. ...
... 本研究中的农田开垦时间仅仅只有短短的5年,但开垦仍然导致土壤微生物群落结构的明显变化,反映出荒漠土壤微生物群落结构对土地利用方式变化的快速响应[8].整体而言,土壤微生物群落结构与植被属性(盖度、生物量和多样性)、土壤EC、TN、TOC、TP和AP显著相关(表3),其强弱关系因微生物类群的不同而异(图5),可能主要由微生物群落的生物学特性所决定.微生物营养假设认为,农业耕作中由于肥料的使用、根系生物量和分泌物等底物的增加,可能会促使一些富营养型微生物相对丰度的增加,而一些贫营养型微生物的丰度降低[30,52].但是,本研究的结果并不支持这一假设.例如,变形菌门和放线菌门往往被认为是富营养型细菌,而酸杆菌门和硝化螺旋菌门(Nitrospirae)则为贫营养型细菌.而此研究中放线菌门、酸杆菌门和硝化螺旋菌门细菌的相对丰度在农田土壤中都显著提高(图3A),这一结果也表明还有其他因素比养分水平在更大程度上决定着因土地利用方式而导致的土壤微生物群落结构的变化,有关这方面的机理还需要进一步的深入研究.同时,最新的一项研究发现,盐分是荒漠土壤细菌和古菌群落结构的关键影响因子,一些耐盐性的细菌和古菌群落存在于盐渍化的荒漠土壤中[14].因此,荒漠在开垦为农田后由于土壤EC的显著减弱,一些耐盐型的微生物,如γ变形菌门(Gammaproteobacteria)细菌和盐杆菌门(Halobacteria)古菌的相对丰度也随之降低.此外,区域尺度[15,53]和控制试验[54]方面的研究发现,水分是影响荒漠土壤微生物群落结构的关键因子.然而,本研究中荒漠和农田土壤的水分没有显著性差异,Mantel test结果也发现土壤水分与微生物群落结构并不相关,表明该区域短期的开垦过程中土壤水分对土壤微生物群落结构的直接作用并不明显,主要是通过影响植被盖度、生物量和土壤EC间接影响土壤微生物的群落构建[15,34].尽管Mantel test分析结果显示土壤TOC和TN仅与细菌群落结构显著正相关,而与古菌和真菌群落结构并不相关(表3),棉田和荒漠土壤之间的TOC和TN也无显著差异(表1),但长期耕作后势必会导致土壤TOC和TN的进一步显著增加[8].众多研究[17,21-23,27]表明长期的施肥和耕作显著改变了土壤微生物群落结构,由此可以预见荒漠农田土壤微生物群落结构可能也势必会进一步发生变化,但由于缺少连续多年的土壤水分、施肥制度和土壤微生物群落变化的相关数据,还无法建立土壤水分动态、施肥制度与土壤微生物群落结构之间的关系.因此,急需通过开展长时间序列的连续动态监测试验进行荒漠土壤微生物群落演替过程及其作用机理方面的研究. ...
荒漠-绿洲土壤微生物群落组成与其活性对比
1
2007
... 土壤微生物通过参与元素生物地球化学和多种生态过程调节影响植物生长、土壤肥力和健康以及生态系统的结构和功能[13].荒漠土壤微生物群落特征与植被群落(生物量、多样性)和土壤特性(养分水平、盐分、pH值、干旱程度等)密切相关[14-15].由于环境对微生物的选择性差异,处于不同生物气候区和不同植被类型的土壤微生物群落特征存在明显的差异[15-16].阿拉尔绿洲的荒漠因具有极干旱、夏季高温、盐渍化土壤、低覆盖度和多样性盐生灌木分布的生物气候特点,可能孕育了一些耐极端条件(如耐干旱和耐盐碱)的土壤微生物群落,从而塑造了有别于其他荒漠的土壤微生物群落结构.在荒漠开垦为农田后,由于作物种植、灌溉、施肥等农业活动,植被结构、土壤养分、盐分和水分等诸多方面均发生了显著变化[17-18],必然导致土壤微生物群落结构的显著改变[19].学者已从耕作方式[20-21]、施肥制度[21-23]、灌溉方式[24-27]等如何影响典型农区土壤微生物群落方面开展了大量研究,但针对干旱区土地利用方式变化如何影响土壤微生物群落的研究相对较少.少数学者采用培养法[28-29]、变性梯度凝胶电泳法(DGGE)[30]、磷脂脂肪酸法(PLFA)[8]、基因测序[17,31-33]、芯片原位杂交[30]等技术对荒漠及其开垦后土壤细菌或真菌群落生物量、多样性及其结构的变化开展了研究,研究区域主要在盐渍化程度不高的干旱或干旱半干旱区,而针对盐渍化、极干旱荒漠在开垦后土壤微生物群落变化的对比研究较少,有关真菌和古菌群落的研究更为稀缺[34],从而限制了我们对荒漠土壤微生物群落如何响应土地利用方式改变的理解和认识. ...
腾格里沙漠东南缘可培养微生物群落数量与结构特征
1
2012
... 土壤微生物通过参与元素生物地球化学和多种生态过程调节影响植物生长、土壤肥力和健康以及生态系统的结构和功能[13].荒漠土壤微生物群落特征与植被群落(生物量、多样性)和土壤特性(养分水平、盐分、pH值、干旱程度等)密切相关[14-15].由于环境对微生物的选择性差异,处于不同生物气候区和不同植被类型的土壤微生物群落特征存在明显的差异[15-16].阿拉尔绿洲的荒漠因具有极干旱、夏季高温、盐渍化土壤、低覆盖度和多样性盐生灌木分布的生物气候特点,可能孕育了一些耐极端条件(如耐干旱和耐盐碱)的土壤微生物群落,从而塑造了有别于其他荒漠的土壤微生物群落结构.在荒漠开垦为农田后,由于作物种植、灌溉、施肥等农业活动,植被结构、土壤养分、盐分和水分等诸多方面均发生了显著变化[17-18],必然导致土壤微生物群落结构的显著改变[19].学者已从耕作方式[20-21]、施肥制度[21-23]、灌溉方式[24-27]等如何影响典型农区土壤微生物群落方面开展了大量研究,但针对干旱区土地利用方式变化如何影响土壤微生物群落的研究相对较少.少数学者采用培养法[28-29]、变性梯度凝胶电泳法(DGGE)[30]、磷脂脂肪酸法(PLFA)[8]、基因测序[17,31-33]、芯片原位杂交[30]等技术对荒漠及其开垦后土壤细菌或真菌群落生物量、多样性及其结构的变化开展了研究,研究区域主要在盐渍化程度不高的干旱或干旱半干旱区,而针对盐渍化、极干旱荒漠在开垦后土壤微生物群落变化的对比研究较少,有关真菌和古菌群落的研究更为稀缺[34],从而限制了我们对荒漠土壤微生物群落如何响应土地利用方式改变的理解和认识. ...
Changes of soil bacterial diversity as a consequence of agricultural land use in a semi-arid ecosystem
4
2013
... 土壤微生物通过参与元素生物地球化学和多种生态过程调节影响植物生长、土壤肥力和健康以及生态系统的结构和功能[13].荒漠土壤微生物群落特征与植被群落(生物量、多样性)和土壤特性(养分水平、盐分、pH值、干旱程度等)密切相关[14-15].由于环境对微生物的选择性差异,处于不同生物气候区和不同植被类型的土壤微生物群落特征存在明显的差异[15-16].阿拉尔绿洲的荒漠因具有极干旱、夏季高温、盐渍化土壤、低覆盖度和多样性盐生灌木分布的生物气候特点,可能孕育了一些耐极端条件(如耐干旱和耐盐碱)的土壤微生物群落,从而塑造了有别于其他荒漠的土壤微生物群落结构.在荒漠开垦为农田后,由于作物种植、灌溉、施肥等农业活动,植被结构、土壤养分、盐分和水分等诸多方面均发生了显著变化[17-18],必然导致土壤微生物群落结构的显著改变[19].学者已从耕作方式[20-21]、施肥制度[21-23]、灌溉方式[24-27]等如何影响典型农区土壤微生物群落方面开展了大量研究,但针对干旱区土地利用方式变化如何影响土壤微生物群落的研究相对较少.少数学者采用培养法[28-29]、变性梯度凝胶电泳法(DGGE)[30]、磷脂脂肪酸法(PLFA)[8]、基因测序[17,31-33]、芯片原位杂交[30]等技术对荒漠及其开垦后土壤细菌或真菌群落生物量、多样性及其结构的变化开展了研究,研究区域主要在盐渍化程度不高的干旱或干旱半干旱区,而针对盐渍化、极干旱荒漠在开垦后土壤微生物群落变化的对比研究较少,有关真菌和古菌群落的研究更为稀缺[34],从而限制了我们对荒漠土壤微生物群落如何响应土地利用方式改变的理解和认识. ...
... [30]等技术对荒漠及其开垦后土壤细菌或真菌群落生物量、多样性及其结构的变化开展了研究,研究区域主要在盐渍化程度不高的干旱或干旱半干旱区,而针对盐渍化、极干旱荒漠在开垦后土壤微生物群落变化的对比研究较少,有关真菌和古菌群落的研究更为稀缺[34],从而限制了我们对荒漠土壤微生物群落如何响应土地利用方式改变的理解和认识. ...
... 天然荒漠开垦为农田后,作物代替了以盐生型灌木为优势种的原生植被,由于耕作、施肥、灌溉和田间管理等因素,导致农田植被盖度和地上生物量显著提高,而植物丰富度则显著下降.与此同时,开垦后土壤理化属性理应也有明显的变化.通常情况下,干旱荒漠在长期耕作(>10年)后,由于植被残体的输入和肥料的使用,表层土壤TOC和TN等养分显著增加,pH值显著下降[8,17-19],而EC显著下降[19,30]或增加[17].本研究发现,荒漠土壤在连续耕作5年后其EC显著下降,TP和AP显著升高,pH值、TOC和TN及其他C、N组分均没有显著变化(表1).这一结果与上述研究报道不太一致,说明开垦对荒漠土壤属性的影响与其所处的生物气候区、灌溉方式、土壤背景、耕作历史等因素密切相关,短期的耕作对极干旱、盐渍化荒漠植被的影响比土壤属性更为明显,对土壤C、N的累积及其有效性和酸碱度影响不大.可能的原因是,阿拉尔绿洲土壤盐分本底值很高,通常分别在冬季和春季采用大水漫灌方式进行盐分的淋洗,在作物生长季以滴灌方式进行灌溉.5年的耕作历史虽短暂,但由于持续的盐分淋洗后表层土壤盐分含量显著降低,导致土壤EC明显下降.值得注意的是,尽管在棉花生产中有N肥的使用和棉花采收后秸秆还田理论上应该会促进土壤TOC和TN水平提升,但本文却发现开垦5年后土壤中TOC和TN及其组分并无显著变化.北疆荒漠开垦为棉田初期(3—5年)土壤TOC和TN的变化也有类似报道,其TOC含量在开垦后的3—5年内甚至降低[8].其可能的原因如下:①开垦初期由于土壤盐分含量高,棉花的地上生物量较低,通过秸秆转化为土壤TOC和TN的数量相对有限,说明开垦后短期的农业耕作还不足以导致该区域土壤C、N等养分含量的显著提升.②大水漫灌导致土壤中的一些可溶性C、N等养分随排水而流失,也很可能导致C、N等养分沿土壤剖面重新分配,导致表层土壤的TOC和TN与天然荒漠无显著差异.③农事耕作和秸秆还田刺激了土壤呼吸,增加了土壤C的排放[37];而灌溉导致农田土壤水分高于荒漠土壤,有利于反硝化作用而导致N素以N2O等气体的形式释放到大气中[38].但是,磷肥的使用还是导致了土壤中TP和AP成分显著富集,从而增加了P的有效性. ...
... 本研究中的农田开垦时间仅仅只有短短的5年,但开垦仍然导致土壤微生物群落结构的明显变化,反映出荒漠土壤微生物群落结构对土地利用方式变化的快速响应[8].整体而言,土壤微生物群落结构与植被属性(盖度、生物量和多样性)、土壤EC、TN、TOC、TP和AP显著相关(表3),其强弱关系因微生物类群的不同而异(图5),可能主要由微生物群落的生物学特性所决定.微生物营养假设认为,农业耕作中由于肥料的使用、根系生物量和分泌物等底物的增加,可能会促使一些富营养型微生物相对丰度的增加,而一些贫营养型微生物的丰度降低[30,52].但是,本研究的结果并不支持这一假设.例如,变形菌门和放线菌门往往被认为是富营养型细菌,而酸杆菌门和硝化螺旋菌门(Nitrospirae)则为贫营养型细菌.而此研究中放线菌门、酸杆菌门和硝化螺旋菌门细菌的相对丰度在农田土壤中都显著提高(图3A),这一结果也表明还有其他因素比养分水平在更大程度上决定着因土地利用方式而导致的土壤微生物群落结构的变化,有关这方面的机理还需要进一步的深入研究.同时,最新的一项研究发现,盐分是荒漠土壤细菌和古菌群落结构的关键影响因子,一些耐盐性的细菌和古菌群落存在于盐渍化的荒漠土壤中[14].因此,荒漠在开垦为农田后由于土壤EC的显著减弱,一些耐盐型的微生物,如γ变形菌门(Gammaproteobacteria)细菌和盐杆菌门(Halobacteria)古菌的相对丰度也随之降低.此外,区域尺度[15,53]和控制试验[54]方面的研究发现,水分是影响荒漠土壤微生物群落结构的关键因子.然而,本研究中荒漠和农田土壤的水分没有显著性差异,Mantel test结果也发现土壤水分与微生物群落结构并不相关,表明该区域短期的开垦过程中土壤水分对土壤微生物群落结构的直接作用并不明显,主要是通过影响植被盖度、生物量和土壤EC间接影响土壤微生物的群落构建[15,34].尽管Mantel test分析结果显示土壤TOC和TN仅与细菌群落结构显著正相关,而与古菌和真菌群落结构并不相关(表3),棉田和荒漠土壤之间的TOC和TN也无显著差异(表1),但长期耕作后势必会导致土壤TOC和TN的进一步显著增加[8].众多研究[17,21-23,27]表明长期的施肥和耕作显著改变了土壤微生物群落结构,由此可以预见荒漠农田土壤微生物群落结构可能也势必会进一步发生变化,但由于缺少连续多年的土壤水分、施肥制度和土壤微生物群落变化的相关数据,还无法建立土壤水分动态、施肥制度与土壤微生物群落结构之间的关系.因此,急需通过开展长时间序列的连续动态监测试验进行荒漠土壤微生物群落演替过程及其作用机理方面的研究. ...
Profile changes in the soil microbial community when desert becomes oasis
2
2015
... 土壤微生物通过参与元素生物地球化学和多种生态过程调节影响植物生长、土壤肥力和健康以及生态系统的结构和功能[13].荒漠土壤微生物群落特征与植被群落(生物量、多样性)和土壤特性(养分水平、盐分、pH值、干旱程度等)密切相关[14-15].由于环境对微生物的选择性差异,处于不同生物气候区和不同植被类型的土壤微生物群落特征存在明显的差异[15-16].阿拉尔绿洲的荒漠因具有极干旱、夏季高温、盐渍化土壤、低覆盖度和多样性盐生灌木分布的生物气候特点,可能孕育了一些耐极端条件(如耐干旱和耐盐碱)的土壤微生物群落,从而塑造了有别于其他荒漠的土壤微生物群落结构.在荒漠开垦为农田后,由于作物种植、灌溉、施肥等农业活动,植被结构、土壤养分、盐分和水分等诸多方面均发生了显著变化[17-18],必然导致土壤微生物群落结构的显著改变[19].学者已从耕作方式[20-21]、施肥制度[21-23]、灌溉方式[24-27]等如何影响典型农区土壤微生物群落方面开展了大量研究,但针对干旱区土地利用方式变化如何影响土壤微生物群落的研究相对较少.少数学者采用培养法[28-29]、变性梯度凝胶电泳法(DGGE)[30]、磷脂脂肪酸法(PLFA)[8]、基因测序[17,31-33]、芯片原位杂交[30]等技术对荒漠及其开垦后土壤细菌或真菌群落生物量、多样性及其结构的变化开展了研究,研究区域主要在盐渍化程度不高的干旱或干旱半干旱区,而针对盐渍化、极干旱荒漠在开垦后土壤微生物群落变化的对比研究较少,有关真菌和古菌群落的研究更为稀缺[34],从而限制了我们对荒漠土壤微生物群落如何响应土地利用方式改变的理解和认识. ...
... 本研究发现,荒漠土壤中变形菌门细菌占绝对优势,其相对丰度(74.8%)远高于其他报道(15.0%—40.0%),放线菌门的相对丰度(0.4%)则远低于其他荒漠地区的报道(23%—-52.0%)[14-15,17,31,46-47];厚壁菌门(Firmicutes)细菌也是干旱荒漠中常见的土壤优势细菌,在非洲的Namib沙漠和中国西部戈壁沙漠土壤中的相对丰度高达50%[48]和80%[47],远远高于本研究中的发现(1.5%);芽单孢菌门细菌的相对丰度(13.1%)明显高于腾格里沙漠土壤(<3.0%)[17].与其他研究类似[15,17],子囊菌门真菌是荒漠土壤中的优势真菌,本研究也发现一些其他未分类真菌门在天然荒漠中也占有较高的相对丰度(图2).本研究中荒漠土壤广古菌门古菌的相对丰度(97.0%)远高于北非的Tataouine 沙漠土壤(40.0%)[45].在Atacama 荒漠、Negev沙漠和内蒙古荒漠草原,土壤真菌群落以曲霉菌属(Aspergillus)和青霉菌属(Penicillium)为主[49-51],而本研究中未分类的真菌属在荒漠土壤真菌群落中占绝对优势(图5C).此外,荒漠土壤中还发现了一些未分类的微生物属(图5),有关这些特殊微生物分类方面的研究还需要进一步加强.可见,盐渍化、极干旱的荒漠环境孕育了有别于其他干旱地区的土壤微生物群落结构. ...
Desert farming benefits from microbial potential in arid soils and promotes diversity and plant health
3
2011
... 一般来讲,土壤水分可利用性和养分水平(如TOC)制约着干旱区土壤微生物群落的生长与繁殖[15,39-40].许多研究发现,土壤细菌和真菌群落生物量与植被盖度和生物量、土壤TOC和TN含量显著正相关[40-41].本研究中,荒漠开垦为农田后其土壤细菌和真菌群落的生物量明显增加,这一研究结果与其他研究报道相一致[8,17,32].主要因为天然荒漠开垦后,绿洲农田植被盖度和生物量显著增加(表1),再加上定期的灌溉,其土壤水热条件得到了极大改善,盐渍化程度大大降低,土壤水分有效性明显增强;同时,农田作物地下根系更为密集,大量的植物残体(秸秆还田)、根系残留物和分泌物输入土壤中,为细菌和真菌群落的生长与繁殖提供了大量的底物.另外,虽然荒漠开垦为农田后,其土壤TN无显著变化,但N和P肥的使用无疑会提高土壤养分的有效性,有利于作物地上和地下生物量的积累,进而促进了细菌和真菌生物量的增加.再者,耕种也增加了表层土壤中的氧气含量,为一些好气型微生物的生理代谢活动创造了更优越的环境.因而,农田土壤细菌和真菌群落生物量明显高于荒漠土壤[8].但是,本研究无法区分耕作、施肥和灌溉对土壤微生物群落特征的各自作用及其机理,有关这方面的研究还需要开展相关的野外控制试验进行深入研究. ...
... 一些原本适应荒漠环境的细菌和真菌群落丰度在开垦为农田后降低或消失[32],或被更能适应农田环境的微生物群落所替代[33].正如我们的预期,荒漠开垦为农田后土壤细菌群落的多样性显著提升,这一结果与之前的研究报道相一致[8,17,32-33].本文的研究也支持了荒漠土壤细菌和古菌群落多样性随着盐分的增加而降低的研究报道[14],由于高盐分的积累提高了细胞外的渗透压,使许多土壤微生物难以适应渗透压胁迫而被迫死亡或休眠[42-43].然而,开垦对荒漠土壤微生物群落的影响因微生物群落的不同而异,荒漠开垦为农田后将有助于提高土壤细菌和真菌群落的生物量,丰富土壤细菌的群落结构.古菌因其具有嗜热、嗜酸碱和嗜盐的特性,往往能适应荒漠干旱、高温、盐渍化的贫营养性环境[44-45],荒漠开垦为农田后因其适宜生境的改变可能导致一些古菌群落的消失或休眠,从而导致其生物量和Shannon多样性的下降.但是,开垦后土壤真菌群落多样性没有明显变化,这一结果与之前的一些研究报道并不一致[8,17,33].由此可见,盐渍化荒漠土壤细菌群落的生物量和多样性响应土壤利用方式的改变可能比真菌和古菌群落更快、更敏感. ...
... ,32-33].本文的研究也支持了荒漠土壤细菌和古菌群落多样性随着盐分的增加而降低的研究报道[14],由于高盐分的积累提高了细胞外的渗透压,使许多土壤微生物难以适应渗透压胁迫而被迫死亡或休眠[42-43].然而,开垦对荒漠土壤微生物群落的影响因微生物群落的不同而异,荒漠开垦为农田后将有助于提高土壤细菌和真菌群落的生物量,丰富土壤细菌的群落结构.古菌因其具有嗜热、嗜酸碱和嗜盐的特性,往往能适应荒漠干旱、高温、盐渍化的贫营养性环境[44-45],荒漠开垦为农田后因其适宜生境的改变可能导致一些古菌群落的消失或休眠,从而导致其生物量和Shannon多样性的下降.但是,开垦后土壤真菌群落多样性没有明显变化,这一结果与之前的一些研究报道并不一致[8,17,33].由此可见,盐渍化荒漠土壤细菌群落的生物量和多样性响应土壤利用方式的改变可能比真菌和古菌群落更快、更敏感. ...
Land-use change alters patterns of soil biodiversity in arid lands of northwestern China
4
2018
... 土壤微生物通过参与元素生物地球化学和多种生态过程调节影响植物生长、土壤肥力和健康以及生态系统的结构和功能[13].荒漠土壤微生物群落特征与植被群落(生物量、多样性)和土壤特性(养分水平、盐分、pH值、干旱程度等)密切相关[14-15].由于环境对微生物的选择性差异,处于不同生物气候区和不同植被类型的土壤微生物群落特征存在明显的差异[15-16].阿拉尔绿洲的荒漠因具有极干旱、夏季高温、盐渍化土壤、低覆盖度和多样性盐生灌木分布的生物气候特点,可能孕育了一些耐极端条件(如耐干旱和耐盐碱)的土壤微生物群落,从而塑造了有别于其他荒漠的土壤微生物群落结构.在荒漠开垦为农田后,由于作物种植、灌溉、施肥等农业活动,植被结构、土壤养分、盐分和水分等诸多方面均发生了显著变化[17-18],必然导致土壤微生物群落结构的显著改变[19].学者已从耕作方式[20-21]、施肥制度[21-23]、灌溉方式[24-27]等如何影响典型农区土壤微生物群落方面开展了大量研究,但针对干旱区土地利用方式变化如何影响土壤微生物群落的研究相对较少.少数学者采用培养法[28-29]、变性梯度凝胶电泳法(DGGE)[30]、磷脂脂肪酸法(PLFA)[8]、基因测序[17,31-33]、芯片原位杂交[30]等技术对荒漠及其开垦后土壤细菌或真菌群落生物量、多样性及其结构的变化开展了研究,研究区域主要在盐渍化程度不高的干旱或干旱半干旱区,而针对盐渍化、极干旱荒漠在开垦后土壤微生物群落变化的对比研究较少,有关真菌和古菌群落的研究更为稀缺[34],从而限制了我们对荒漠土壤微生物群落如何响应土地利用方式改变的理解和认识. ...
... 一些原本适应荒漠环境的细菌和真菌群落丰度在开垦为农田后降低或消失[32],或被更能适应农田环境的微生物群落所替代[33].正如我们的预期,荒漠开垦为农田后土壤细菌群落的多样性显著提升,这一结果与之前的研究报道相一致[8,17,32-33].本文的研究也支持了荒漠土壤细菌和古菌群落多样性随着盐分的增加而降低的研究报道[14],由于高盐分的积累提高了细胞外的渗透压,使许多土壤微生物难以适应渗透压胁迫而被迫死亡或休眠[42-43].然而,开垦对荒漠土壤微生物群落的影响因微生物群落的不同而异,荒漠开垦为农田后将有助于提高土壤细菌和真菌群落的生物量,丰富土壤细菌的群落结构.古菌因其具有嗜热、嗜酸碱和嗜盐的特性,往往能适应荒漠干旱、高温、盐渍化的贫营养性环境[44-45],荒漠开垦为农田后因其适宜生境的改变可能导致一些古菌群落的消失或休眠,从而导致其生物量和Shannon多样性的下降.但是,开垦后土壤真菌群落多样性没有明显变化,这一结果与之前的一些研究报道并不一致[8,17,33].由此可见,盐渍化荒漠土壤细菌群落的生物量和多样性响应土壤利用方式的改变可能比真菌和古菌群落更快、更敏感. ...
... -33].本文的研究也支持了荒漠土壤细菌和古菌群落多样性随着盐分的增加而降低的研究报道[14],由于高盐分的积累提高了细胞外的渗透压,使许多土壤微生物难以适应渗透压胁迫而被迫死亡或休眠[42-43].然而,开垦对荒漠土壤微生物群落的影响因微生物群落的不同而异,荒漠开垦为农田后将有助于提高土壤细菌和真菌群落的生物量,丰富土壤细菌的群落结构.古菌因其具有嗜热、嗜酸碱和嗜盐的特性,往往能适应荒漠干旱、高温、盐渍化的贫营养性环境[44-45],荒漠开垦为农田后因其适宜生境的改变可能导致一些古菌群落的消失或休眠,从而导致其生物量和Shannon多样性的下降.但是,开垦后土壤真菌群落多样性没有明显变化,这一结果与之前的一些研究报道并不一致[8,17,33].由此可见,盐渍化荒漠土壤细菌群落的生物量和多样性响应土壤利用方式的改变可能比真菌和古菌群落更快、更敏感. ...
... ,33].由此可见,盐渍化荒漠土壤细菌群落的生物量和多样性响应土壤利用方式的改变可能比真菌和古菌群落更快、更敏感. ...
荒漠土壤微生物群落结构特征研究进展
2
2018
... 土壤微生物通过参与元素生物地球化学和多种生态过程调节影响植物生长、土壤肥力和健康以及生态系统的结构和功能[13].荒漠土壤微生物群落特征与植被群落(生物量、多样性)和土壤特性(养分水平、盐分、pH值、干旱程度等)密切相关[14-15].由于环境对微生物的选择性差异,处于不同生物气候区和不同植被类型的土壤微生物群落特征存在明显的差异[15-16].阿拉尔绿洲的荒漠因具有极干旱、夏季高温、盐渍化土壤、低覆盖度和多样性盐生灌木分布的生物气候特点,可能孕育了一些耐极端条件(如耐干旱和耐盐碱)的土壤微生物群落,从而塑造了有别于其他荒漠的土壤微生物群落结构.在荒漠开垦为农田后,由于作物种植、灌溉、施肥等农业活动,植被结构、土壤养分、盐分和水分等诸多方面均发生了显著变化[17-18],必然导致土壤微生物群落结构的显著改变[19].学者已从耕作方式[20-21]、施肥制度[21-23]、灌溉方式[24-27]等如何影响典型农区土壤微生物群落方面开展了大量研究,但针对干旱区土地利用方式变化如何影响土壤微生物群落的研究相对较少.少数学者采用培养法[28-29]、变性梯度凝胶电泳法(DGGE)[30]、磷脂脂肪酸法(PLFA)[8]、基因测序[17,31-33]、芯片原位杂交[30]等技术对荒漠及其开垦后土壤细菌或真菌群落生物量、多样性及其结构的变化开展了研究,研究区域主要在盐渍化程度不高的干旱或干旱半干旱区,而针对盐渍化、极干旱荒漠在开垦后土壤微生物群落变化的对比研究较少,有关真菌和古菌群落的研究更为稀缺[34],从而限制了我们对荒漠土壤微生物群落如何响应土地利用方式改变的理解和认识. ...
... 本研究中的农田开垦时间仅仅只有短短的5年,但开垦仍然导致土壤微生物群落结构的明显变化,反映出荒漠土壤微生物群落结构对土地利用方式变化的快速响应[8].整体而言,土壤微生物群落结构与植被属性(盖度、生物量和多样性)、土壤EC、TN、TOC、TP和AP显著相关(表3),其强弱关系因微生物类群的不同而异(图5),可能主要由微生物群落的生物学特性所决定.微生物营养假设认为,农业耕作中由于肥料的使用、根系生物量和分泌物等底物的增加,可能会促使一些富营养型微生物相对丰度的增加,而一些贫营养型微生物的丰度降低[30,52].但是,本研究的结果并不支持这一假设.例如,变形菌门和放线菌门往往被认为是富营养型细菌,而酸杆菌门和硝化螺旋菌门(Nitrospirae)则为贫营养型细菌.而此研究中放线菌门、酸杆菌门和硝化螺旋菌门细菌的相对丰度在农田土壤中都显著提高(图3A),这一结果也表明还有其他因素比养分水平在更大程度上决定着因土地利用方式而导致的土壤微生物群落结构的变化,有关这方面的机理还需要进一步的深入研究.同时,最新的一项研究发现,盐分是荒漠土壤细菌和古菌群落结构的关键影响因子,一些耐盐性的细菌和古菌群落存在于盐渍化的荒漠土壤中[14].因此,荒漠在开垦为农田后由于土壤EC的显著减弱,一些耐盐型的微生物,如γ变形菌门(Gammaproteobacteria)细菌和盐杆菌门(Halobacteria)古菌的相对丰度也随之降低.此外,区域尺度[15,53]和控制试验[54]方面的研究发现,水分是影响荒漠土壤微生物群落结构的关键因子.然而,本研究中荒漠和农田土壤的水分没有显著性差异,Mantel test结果也发现土壤水分与微生物群落结构并不相关,表明该区域短期的开垦过程中土壤水分对土壤微生物群落结构的直接作用并不明显,主要是通过影响植被盖度、生物量和土壤EC间接影响土壤微生物的群落构建[15,34].尽管Mantel test分析结果显示土壤TOC和TN仅与细菌群落结构显著正相关,而与古菌和真菌群落结构并不相关(表3),棉田和荒漠土壤之间的TOC和TN也无显著差异(表1),但长期耕作后势必会导致土壤TOC和TN的进一步显著增加[8].众多研究[17,21-23,27]表明长期的施肥和耕作显著改变了土壤微生物群落结构,由此可以预见荒漠农田土壤微生物群落结构可能也势必会进一步发生变化,但由于缺少连续多年的土壤水分、施肥制度和土壤微生物群落变化的相关数据,还无法建立土壤水分动态、施肥制度与土壤微生物群落结构之间的关系.因此,急需通过开展长时间序列的连续动态监测试验进行荒漠土壤微生物群落演替过程及其作用机理方面的研究. ...
Configuration of water resources for a typical river basin in an arid region of China based on the ecological water requirements (EWRs) of desert riparian vegetation
1
2014
... 试验地位于新疆的阿拉尔绿洲垦区边缘(40°44′09″N、81°08′10″E,海拔974 m).该区域是阿克苏河、和田河和叶尔羌河交汇之地,属于暖温带极端干旱气候区,区内广泛分布着盐碱型天然荒漠和人工开垦的农田.多年平均气温11.6 ℃,年降水量45.7 mm,降水多在6—10月,年日照2 940 h,年蒸发量超过2 500 mm.土壤为沙质潮土,土壤盐渍化程度高.天然植被以盐生灌木为主,主要优势物种有盐穗木(Halostachys caspica)、柽柳(Tamarix ramosissima)、梭梭(Haloxylon ammodendron)、小果白刺(Nitraria sibirica)等,草本以骆驼刺(Alhagi sparsifolia)、芦苇(Phragmites australis)、胀果甘草(Glyzyrrhiza inflata)和花花柴(Kare-linia caspica)为主[35].农田以连续耕作种植棉花为主. ...
Experimental evaluation of methods to quantify dissolved organic nitrogen (DON) and dissolved organic carbon (DOC) in soil
1
2006
... 土壤含水量(SWC)用烘干称重法(105 ℃)测定;pH值和电导率(EC)分别采用电位法和电导法(土∶水=1∶2.5)测定;采用Costech元素分析仪(ECS 4010, Valencia, 美国)测定土壤总有机碳(TOC)和全氮(TN);全磷(TP)采用酸溶-钼锑抗比色法测定;土壤有效磷(AP)采用NaHCO3浸提-钼锑抗比色法测定(Olsen and Sommers,1982).用2 mol·L-1的KCl(土∶水=1∶2.5)提取土壤中的DOC、DON、铵态氮(Nitrate)和硝态氮(Ammonia)[36],在TOC/TN分析仪(Vario TOC, Elementar,德国)和连续流分析仪(Skalar San++,荷兰)上进行分析. ...
秸秆还田对绿洲棉田土壤CO2时空分布的影响
1
2017
... 天然荒漠开垦为农田后,作物代替了以盐生型灌木为优势种的原生植被,由于耕作、施肥、灌溉和田间管理等因素,导致农田植被盖度和地上生物量显著提高,而植物丰富度则显著下降.与此同时,开垦后土壤理化属性理应也有明显的变化.通常情况下,干旱荒漠在长期耕作(>10年)后,由于植被残体的输入和肥料的使用,表层土壤TOC和TN等养分显著增加,pH值显著下降[8,17-19],而EC显著下降[19,30]或增加[17].本研究发现,荒漠土壤在连续耕作5年后其EC显著下降,TP和AP显著升高,pH值、TOC和TN及其他C、N组分均没有显著变化(表1).这一结果与上述研究报道不太一致,说明开垦对荒漠土壤属性的影响与其所处的生物气候区、灌溉方式、土壤背景、耕作历史等因素密切相关,短期的耕作对极干旱、盐渍化荒漠植被的影响比土壤属性更为明显,对土壤C、N的累积及其有效性和酸碱度影响不大.可能的原因是,阿拉尔绿洲土壤盐分本底值很高,通常分别在冬季和春季采用大水漫灌方式进行盐分的淋洗,在作物生长季以滴灌方式进行灌溉.5年的耕作历史虽短暂,但由于持续的盐分淋洗后表层土壤盐分含量显著降低,导致土壤EC明显下降.值得注意的是,尽管在棉花生产中有N肥的使用和棉花采收后秸秆还田理论上应该会促进土壤TOC和TN水平提升,但本文却发现开垦5年后土壤中TOC和TN及其组分并无显著变化.北疆荒漠开垦为棉田初期(3—5年)土壤TOC和TN的变化也有类似报道,其TOC含量在开垦后的3—5年内甚至降低[8].其可能的原因如下:①开垦初期由于土壤盐分含量高,棉花的地上生物量较低,通过秸秆转化为土壤TOC和TN的数量相对有限,说明开垦后短期的农业耕作还不足以导致该区域土壤C、N等养分含量的显著提升.②大水漫灌导致土壤中的一些可溶性C、N等养分随排水而流失,也很可能导致C、N等养分沿土壤剖面重新分配,导致表层土壤的TOC和TN与天然荒漠无显著差异.③农事耕作和秸秆还田刺激了土壤呼吸,增加了土壤C的排放[37];而灌溉导致农田土壤水分高于荒漠土壤,有利于反硝化作用而导致N素以N2O等气体的形式释放到大气中[38].但是,磷肥的使用还是导致了土壤中TP和AP成分显著富集,从而增加了P的有效性. ...
不同水肥条件对绿洲农田土壤N2O排放的影响
1
2018
... 天然荒漠开垦为农田后,作物代替了以盐生型灌木为优势种的原生植被,由于耕作、施肥、灌溉和田间管理等因素,导致农田植被盖度和地上生物量显著提高,而植物丰富度则显著下降.与此同时,开垦后土壤理化属性理应也有明显的变化.通常情况下,干旱荒漠在长期耕作(>10年)后,由于植被残体的输入和肥料的使用,表层土壤TOC和TN等养分显著增加,pH值显著下降[8,17-19],而EC显著下降[19,30]或增加[17].本研究发现,荒漠土壤在连续耕作5年后其EC显著下降,TP和AP显著升高,pH值、TOC和TN及其他C、N组分均没有显著变化(表1).这一结果与上述研究报道不太一致,说明开垦对荒漠土壤属性的影响与其所处的生物气候区、灌溉方式、土壤背景、耕作历史等因素密切相关,短期的耕作对极干旱、盐渍化荒漠植被的影响比土壤属性更为明显,对土壤C、N的累积及其有效性和酸碱度影响不大.可能的原因是,阿拉尔绿洲土壤盐分本底值很高,通常分别在冬季和春季采用大水漫灌方式进行盐分的淋洗,在作物生长季以滴灌方式进行灌溉.5年的耕作历史虽短暂,但由于持续的盐分淋洗后表层土壤盐分含量显著降低,导致土壤EC明显下降.值得注意的是,尽管在棉花生产中有N肥的使用和棉花采收后秸秆还田理论上应该会促进土壤TOC和TN水平提升,但本文却发现开垦5年后土壤中TOC和TN及其组分并无显著变化.北疆荒漠开垦为棉田初期(3—5年)土壤TOC和TN的变化也有类似报道,其TOC含量在开垦后的3—5年内甚至降低[8].其可能的原因如下:①开垦初期由于土壤盐分含量高,棉花的地上生物量较低,通过秸秆转化为土壤TOC和TN的数量相对有限,说明开垦后短期的农业耕作还不足以导致该区域土壤C、N等养分含量的显著提升.②大水漫灌导致土壤中的一些可溶性C、N等养分随排水而流失,也很可能导致C、N等养分沿土壤剖面重新分配,导致表层土壤的TOC和TN与天然荒漠无显著差异.③农事耕作和秸秆还田刺激了土壤呼吸,增加了土壤C的排放[37];而灌溉导致农田土壤水分高于荒漠土壤,有利于反硝化作用而导致N素以N2O等气体的形式释放到大气中[38].但是,磷肥的使用还是导致了土壤中TP和AP成分显著富集,从而增加了P的有效性. ...
Active and total prokaryotic communities in dryland soils
1
2013
... 一般来讲,土壤水分可利用性和养分水平(如TOC)制约着干旱区土壤微生物群落的生长与繁殖[15,39-40].许多研究发现,土壤细菌和真菌群落生物量与植被盖度和生物量、土壤TOC和TN含量显著正相关[40-41].本研究中,荒漠开垦为农田后其土壤细菌和真菌群落的生物量明显增加,这一研究结果与其他研究报道相一致[8,17,32].主要因为天然荒漠开垦后,绿洲农田植被盖度和生物量显著增加(表1),再加上定期的灌溉,其土壤水热条件得到了极大改善,盐渍化程度大大降低,土壤水分有效性明显增强;同时,农田作物地下根系更为密集,大量的植物残体(秸秆还田)、根系残留物和分泌物输入土壤中,为细菌和真菌群落的生长与繁殖提供了大量的底物.另外,虽然荒漠开垦为农田后,其土壤TN无显著变化,但N和P肥的使用无疑会提高土壤养分的有效性,有利于作物地上和地下生物量的积累,进而促进了细菌和真菌生物量的增加.再者,耕种也增加了表层土壤中的氧气含量,为一些好气型微生物的生理代谢活动创造了更优越的环境.因而,农田土壤细菌和真菌群落生物量明显高于荒漠土壤[8].但是,本研究无法区分耕作、施肥和灌溉对土壤微生物群落特征的各自作用及其机理,有关这方面的研究还需要开展相关的野外控制试验进行深入研究. ...
Distribution and abundance of soil fungi in Antarctica at sites on the Peninsula,Ross Sea Region and McMurdo Dry Valleys
2
2011
... 一般来讲,土壤水分可利用性和养分水平(如TOC)制约着干旱区土壤微生物群落的生长与繁殖[15,39-40].许多研究发现,土壤细菌和真菌群落生物量与植被盖度和生物量、土壤TOC和TN含量显著正相关[40-41].本研究中,荒漠开垦为农田后其土壤细菌和真菌群落的生物量明显增加,这一研究结果与其他研究报道相一致[8,17,32].主要因为天然荒漠开垦后,绿洲农田植被盖度和生物量显著增加(表1),再加上定期的灌溉,其土壤水热条件得到了极大改善,盐渍化程度大大降低,土壤水分有效性明显增强;同时,农田作物地下根系更为密集,大量的植物残体(秸秆还田)、根系残留物和分泌物输入土壤中,为细菌和真菌群落的生长与繁殖提供了大量的底物.另外,虽然荒漠开垦为农田后,其土壤TN无显著变化,但N和P肥的使用无疑会提高土壤养分的有效性,有利于作物地上和地下生物量的积累,进而促进了细菌和真菌生物量的增加.再者,耕种也增加了表层土壤中的氧气含量,为一些好气型微生物的生理代谢活动创造了更优越的环境.因而,农田土壤细菌和真菌群落生物量明显高于荒漠土壤[8].但是,本研究无法区分耕作、施肥和灌溉对土壤微生物群落特征的各自作用及其机理,有关这方面的研究还需要开展相关的野外控制试验进行深入研究. ...
... [40-41].本研究中,荒漠开垦为农田后其土壤细菌和真菌群落的生物量明显增加,这一研究结果与其他研究报道相一致[8,17,32].主要因为天然荒漠开垦后,绿洲农田植被盖度和生物量显著增加(表1),再加上定期的灌溉,其土壤水热条件得到了极大改善,盐渍化程度大大降低,土壤水分有效性明显增强;同时,农田作物地下根系更为密集,大量的植物残体(秸秆还田)、根系残留物和分泌物输入土壤中,为细菌和真菌群落的生长与繁殖提供了大量的底物.另外,虽然荒漠开垦为农田后,其土壤TN无显著变化,但N和P肥的使用无疑会提高土壤养分的有效性,有利于作物地上和地下生物量的积累,进而促进了细菌和真菌生物量的增加.再者,耕种也增加了表层土壤中的氧气含量,为一些好气型微生物的生理代谢活动创造了更优越的环境.因而,农田土壤细菌和真菌群落生物量明显高于荒漠土壤[8].但是,本研究无法区分耕作、施肥和灌溉对土壤微生物群落特征的各自作用及其机理,有关这方面的研究还需要开展相关的野外控制试验进行深入研究. ...
Shifts in soil microbial community functional gene structure across a 61-year desert revegetation chronosequence
1
2019
... 一般来讲,土壤水分可利用性和养分水平(如TOC)制约着干旱区土壤微生物群落的生长与繁殖[15,39-40].许多研究发现,土壤细菌和真菌群落生物量与植被盖度和生物量、土壤TOC和TN含量显著正相关[40-41].本研究中,荒漠开垦为农田后其土壤细菌和真菌群落的生物量明显增加,这一研究结果与其他研究报道相一致[8,17,32].主要因为天然荒漠开垦后,绿洲农田植被盖度和生物量显著增加(表1),再加上定期的灌溉,其土壤水热条件得到了极大改善,盐渍化程度大大降低,土壤水分有效性明显增强;同时,农田作物地下根系更为密集,大量的植物残体(秸秆还田)、根系残留物和分泌物输入土壤中,为细菌和真菌群落的生长与繁殖提供了大量的底物.另外,虽然荒漠开垦为农田后,其土壤TN无显著变化,但N和P肥的使用无疑会提高土壤养分的有效性,有利于作物地上和地下生物量的积累,进而促进了细菌和真菌生物量的增加.再者,耕种也增加了表层土壤中的氧气含量,为一些好气型微生物的生理代谢活动创造了更优越的环境.因而,农田土壤细菌和真菌群落生物量明显高于荒漠土壤[8].但是,本研究无法区分耕作、施肥和灌溉对土壤微生物群落特征的各自作用及其机理,有关这方面的研究还需要开展相关的野外控制试验进行深入研究. ...
Salt effects on the soil microbial decomposer community and their role in organic carbon cycling:a review
1
2015
... 一些原本适应荒漠环境的细菌和真菌群落丰度在开垦为农田后降低或消失[32],或被更能适应农田环境的微生物群落所替代[33].正如我们的预期,荒漠开垦为农田后土壤细菌群落的多样性显著提升,这一结果与之前的研究报道相一致[8,17,32-33].本文的研究也支持了荒漠土壤细菌和古菌群落多样性随着盐分的增加而降低的研究报道[14],由于高盐分的积累提高了细胞外的渗透压,使许多土壤微生物难以适应渗透压胁迫而被迫死亡或休眠[42-43].然而,开垦对荒漠土壤微生物群落的影响因微生物群落的不同而异,荒漠开垦为农田后将有助于提高土壤细菌和真菌群落的生物量,丰富土壤细菌的群落结构.古菌因其具有嗜热、嗜酸碱和嗜盐的特性,往往能适应荒漠干旱、高温、盐渍化的贫营养性环境[44-45],荒漠开垦为农田后因其适宜生境的改变可能导致一些古菌群落的消失或休眠,从而导致其生物量和Shannon多样性的下降.但是,开垦后土壤真菌群落多样性没有明显变化,这一结果与之前的一些研究报道并不一致[8,17,33].由此可见,盐渍化荒漠土壤细菌群落的生物量和多样性响应土壤利用方式的改变可能比真菌和古菌群落更快、更敏感. ...
Thermodynamic limits to microbial life at high salt concentrations
1
2011
... 一些原本适应荒漠环境的细菌和真菌群落丰度在开垦为农田后降低或消失[32],或被更能适应农田环境的微生物群落所替代[33].正如我们的预期,荒漠开垦为农田后土壤细菌群落的多样性显著提升,这一结果与之前的研究报道相一致[8,17,32-33].本文的研究也支持了荒漠土壤细菌和古菌群落多样性随着盐分的增加而降低的研究报道[14],由于高盐分的积累提高了细胞外的渗透压,使许多土壤微生物难以适应渗透压胁迫而被迫死亡或休眠[42-43].然而,开垦对荒漠土壤微生物群落的影响因微生物群落的不同而异,荒漠开垦为农田后将有助于提高土壤细菌和真菌群落的生物量,丰富土壤细菌的群落结构.古菌因其具有嗜热、嗜酸碱和嗜盐的特性,往往能适应荒漠干旱、高温、盐渍化的贫营养性环境[44-45],荒漠开垦为农田后因其适宜生境的改变可能导致一些古菌群落的消失或休眠,从而导致其生物量和Shannon多样性的下降.但是,开垦后土壤真菌群落多样性没有明显变化,这一结果与之前的一些研究报道并不一致[8,17,33].由此可见,盐渍化荒漠土壤细菌群落的生物量和多样性响应土壤利用方式的改变可能比真菌和古菌群落更快、更敏感. ...
Microflora of extreme arid Atacama Desert soils
1
2007
... 一些原本适应荒漠环境的细菌和真菌群落丰度在开垦为农田后降低或消失[32],或被更能适应农田环境的微生物群落所替代[33].正如我们的预期,荒漠开垦为农田后土壤细菌群落的多样性显著提升,这一结果与之前的研究报道相一致[8,17,32-33].本文的研究也支持了荒漠土壤细菌和古菌群落多样性随着盐分的增加而降低的研究报道[14],由于高盐分的积累提高了细胞外的渗透压,使许多土壤微生物难以适应渗透压胁迫而被迫死亡或休眠[42-43].然而,开垦对荒漠土壤微生物群落的影响因微生物群落的不同而异,荒漠开垦为农田后将有助于提高土壤细菌和真菌群落的生物量,丰富土壤细菌的群落结构.古菌因其具有嗜热、嗜酸碱和嗜盐的特性,往往能适应荒漠干旱、高温、盐渍化的贫营养性环境[44-45],荒漠开垦为农田后因其适宜生境的改变可能导致一些古菌群落的消失或休眠,从而导致其生物量和Shannon多样性的下降.但是,开垦后土壤真菌群落多样性没有明显变化,这一结果与之前的一些研究报道并不一致[8,17,33].由此可见,盐渍化荒漠土壤细菌群落的生物量和多样性响应土壤利用方式的改变可能比真菌和古菌群落更快、更敏感. ...
The desert of Tataouine:an extreme environment that hosts a wide diversity of microorganisms and radiotolerant bacteria
2
2006
... 一些原本适应荒漠环境的细菌和真菌群落丰度在开垦为农田后降低或消失[32],或被更能适应农田环境的微生物群落所替代[33].正如我们的预期,荒漠开垦为农田后土壤细菌群落的多样性显著提升,这一结果与之前的研究报道相一致[8,17,32-33].本文的研究也支持了荒漠土壤细菌和古菌群落多样性随着盐分的增加而降低的研究报道[14],由于高盐分的积累提高了细胞外的渗透压,使许多土壤微生物难以适应渗透压胁迫而被迫死亡或休眠[42-43].然而,开垦对荒漠土壤微生物群落的影响因微生物群落的不同而异,荒漠开垦为农田后将有助于提高土壤细菌和真菌群落的生物量,丰富土壤细菌的群落结构.古菌因其具有嗜热、嗜酸碱和嗜盐的特性,往往能适应荒漠干旱、高温、盐渍化的贫营养性环境[44-45],荒漠开垦为农田后因其适宜生境的改变可能导致一些古菌群落的消失或休眠,从而导致其生物量和Shannon多样性的下降.但是,开垦后土壤真菌群落多样性没有明显变化,这一结果与之前的一些研究报道并不一致[8,17,33].由此可见,盐渍化荒漠土壤细菌群落的生物量和多样性响应土壤利用方式的改变可能比真菌和古菌群落更快、更敏感. ...
... 本研究发现,荒漠土壤中变形菌门细菌占绝对优势,其相对丰度(74.8%)远高于其他报道(15.0%—40.0%),放线菌门的相对丰度(0.4%)则远低于其他荒漠地区的报道(23%—-52.0%)[14-15,17,31,46-47];厚壁菌门(Firmicutes)细菌也是干旱荒漠中常见的土壤优势细菌,在非洲的Namib沙漠和中国西部戈壁沙漠土壤中的相对丰度高达50%[48]和80%[47],远远高于本研究中的发现(1.5%);芽单孢菌门细菌的相对丰度(13.1%)明显高于腾格里沙漠土壤(<3.0%)[17].与其他研究类似[15,17],子囊菌门真菌是荒漠土壤中的优势真菌,本研究也发现一些其他未分类真菌门在天然荒漠中也占有较高的相对丰度(图2).本研究中荒漠土壤广古菌门古菌的相对丰度(97.0%)远高于北非的Tataouine 沙漠土壤(40.0%)[45].在Atacama 荒漠、Negev沙漠和内蒙古荒漠草原,土壤真菌群落以曲霉菌属(Aspergillus)和青霉菌属(Penicillium)为主[49-51],而本研究中未分类的真菌属在荒漠土壤真菌群落中占绝对优势(图5C).此外,荒漠土壤中还发现了一些未分类的微生物属(图5),有关这些特殊微生物分类方面的研究还需要进一步加强.可见,盐渍化、极干旱的荒漠环境孕育了有别于其他干旱地区的土壤微生物群落结构. ...
Microbial diversity in desert ecosystems
1
2005
... 本研究发现,荒漠土壤中变形菌门细菌占绝对优势,其相对丰度(74.8%)远高于其他报道(15.0%—40.0%),放线菌门的相对丰度(0.4%)则远低于其他荒漠地区的报道(23%—-52.0%)[14-15,17,31,46-47];厚壁菌门(Firmicutes)细菌也是干旱荒漠中常见的土壤优势细菌,在非洲的Namib沙漠和中国西部戈壁沙漠土壤中的相对丰度高达50%[48]和80%[47],远远高于本研究中的发现(1.5%);芽单孢菌门细菌的相对丰度(13.1%)明显高于腾格里沙漠土壤(<3.0%)[17].与其他研究类似[15,17],子囊菌门真菌是荒漠土壤中的优势真菌,本研究也发现一些其他未分类真菌门在天然荒漠中也占有较高的相对丰度(图2).本研究中荒漠土壤广古菌门古菌的相对丰度(97.0%)远高于北非的Tataouine 沙漠土壤(40.0%)[45].在Atacama 荒漠、Negev沙漠和内蒙古荒漠草原,土壤真菌群落以曲霉菌属(Aspergillus)和青霉菌属(Penicillium)为主[49-51],而本研究中未分类的真菌属在荒漠土壤真菌群落中占绝对优势(图5C).此外,荒漠土壤中还发现了一些未分类的微生物属(图5),有关这些特殊微生物分类方面的研究还需要进一步加强.可见,盐渍化、极干旱的荒漠环境孕育了有别于其他干旱地区的土壤微生物群落结构. ...
Bacterial diversity of surface sand samples from the Gobi and Taklamaken Deserts
2
2013
... 本研究发现,荒漠土壤中变形菌门细菌占绝对优势,其相对丰度(74.8%)远高于其他报道(15.0%—40.0%),放线菌门的相对丰度(0.4%)则远低于其他荒漠地区的报道(23%—-52.0%)[14-15,17,31,46-47];厚壁菌门(Firmicutes)细菌也是干旱荒漠中常见的土壤优势细菌,在非洲的Namib沙漠和中国西部戈壁沙漠土壤中的相对丰度高达50%[48]和80%[47],远远高于本研究中的发现(1.5%);芽单孢菌门细菌的相对丰度(13.1%)明显高于腾格里沙漠土壤(<3.0%)[17].与其他研究类似[15,17],子囊菌门真菌是荒漠土壤中的优势真菌,本研究也发现一些其他未分类真菌门在天然荒漠中也占有较高的相对丰度(图2).本研究中荒漠土壤广古菌门古菌的相对丰度(97.0%)远高于北非的Tataouine 沙漠土壤(40.0%)[45].在Atacama 荒漠、Negev沙漠和内蒙古荒漠草原,土壤真菌群落以曲霉菌属(Aspergillus)和青霉菌属(Penicillium)为主[49-51],而本研究中未分类的真菌属在荒漠土壤真菌群落中占绝对优势(图5C).此外,荒漠土壤中还发现了一些未分类的微生物属(图5),有关这些特殊微生物分类方面的研究还需要进一步加强.可见,盐渍化、极干旱的荒漠环境孕育了有别于其他干旱地区的土壤微生物群落结构. ...
... [47],远远高于本研究中的发现(1.5%);芽单孢菌门细菌的相对丰度(13.1%)明显高于腾格里沙漠土壤(<3.0%)[17].与其他研究类似[15,17],子囊菌门真菌是荒漠土壤中的优势真菌,本研究也发现一些其他未分类真菌门在天然荒漠中也占有较高的相对丰度(图2).本研究中荒漠土壤广古菌门古菌的相对丰度(97.0%)远高于北非的Tataouine 沙漠土壤(40.0%)[45].在Atacama 荒漠、Negev沙漠和内蒙古荒漠草原,土壤真菌群落以曲霉菌属(Aspergillus)和青霉菌属(Penicillium)为主[49-51],而本研究中未分类的真菌属在荒漠土壤真菌群落中占绝对优势(图5C).此外,荒漠土壤中还发现了一些未分类的微生物属(图5),有关这些特殊微生物分类方面的研究还需要进一步加强.可见,盐渍化、极干旱的荒漠环境孕育了有别于其他干旱地区的土壤微生物群落结构. ...
An examination of the bacteriophages and bacteria of the Namib desert
1
2008
... 本研究发现,荒漠土壤中变形菌门细菌占绝对优势,其相对丰度(74.8%)远高于其他报道(15.0%—40.0%),放线菌门的相对丰度(0.4%)则远低于其他荒漠地区的报道(23%—-52.0%)[14-15,17,31,46-47];厚壁菌门(Firmicutes)细菌也是干旱荒漠中常见的土壤优势细菌,在非洲的Namib沙漠和中国西部戈壁沙漠土壤中的相对丰度高达50%[48]和80%[47],远远高于本研究中的发现(1.5%);芽单孢菌门细菌的相对丰度(13.1%)明显高于腾格里沙漠土壤(<3.0%)[17].与其他研究类似[15,17],子囊菌门真菌是荒漠土壤中的优势真菌,本研究也发现一些其他未分类真菌门在天然荒漠中也占有较高的相对丰度(图2).本研究中荒漠土壤广古菌门古菌的相对丰度(97.0%)远高于北非的Tataouine 沙漠土壤(40.0%)[45].在Atacama 荒漠、Negev沙漠和内蒙古荒漠草原,土壤真菌群落以曲霉菌属(Aspergillus)和青霉菌属(Penicillium)为主[49-51],而本研究中未分类的真菌属在荒漠土壤真菌群落中占绝对优势(图5C).此外,荒漠土壤中还发现了一些未分类的微生物属(图5),有关这些特殊微生物分类方面的研究还需要进一步加强.可见,盐渍化、极干旱的荒漠环境孕育了有别于其他干旱地区的土壤微生物群落结构. ...
Fungi associated with rocks of the Atacama Desert:taxonomy,distribution,diversity,ecology and bioprospection for bioactive compounds
1
2016
... 本研究发现,荒漠土壤中变形菌门细菌占绝对优势,其相对丰度(74.8%)远高于其他报道(15.0%—40.0%),放线菌门的相对丰度(0.4%)则远低于其他荒漠地区的报道(23%—-52.0%)[14-15,17,31,46-47];厚壁菌门(Firmicutes)细菌也是干旱荒漠中常见的土壤优势细菌,在非洲的Namib沙漠和中国西部戈壁沙漠土壤中的相对丰度高达50%[48]和80%[47],远远高于本研究中的发现(1.5%);芽单孢菌门细菌的相对丰度(13.1%)明显高于腾格里沙漠土壤(<3.0%)[17].与其他研究类似[15,17],子囊菌门真菌是荒漠土壤中的优势真菌,本研究也发现一些其他未分类真菌门在天然荒漠中也占有较高的相对丰度(图2).本研究中荒漠土壤广古菌门古菌的相对丰度(97.0%)远高于北非的Tataouine 沙漠土壤(40.0%)[45].在Atacama 荒漠、Negev沙漠和内蒙古荒漠草原,土壤真菌群落以曲霉菌属(Aspergillus)和青霉菌属(Penicillium)为主[49-51],而本研究中未分类的真菌属在荒漠土壤真菌群落中占绝对优势(图5C).此外,荒漠土壤中还发现了一些未分类的微生物属(图5),有关这些特殊微生物分类方面的研究还需要进一步加强.可见,盐渍化、极干旱的荒漠环境孕育了有别于其他干旱地区的土壤微生物群落结构. ...
Spatiotemporal distribution of soil microfungi in the Makhtesh Ramon area,central Negev Desert,Israel
2010
内蒙古荒漠草原土壤可培养真菌的群落结构和空间分布分析
1
2017
... 本研究发现,荒漠土壤中变形菌门细菌占绝对优势,其相对丰度(74.8%)远高于其他报道(15.0%—40.0%),放线菌门的相对丰度(0.4%)则远低于其他荒漠地区的报道(23%—-52.0%)[14-15,17,31,46-47];厚壁菌门(Firmicutes)细菌也是干旱荒漠中常见的土壤优势细菌,在非洲的Namib沙漠和中国西部戈壁沙漠土壤中的相对丰度高达50%[48]和80%[47],远远高于本研究中的发现(1.5%);芽单孢菌门细菌的相对丰度(13.1%)明显高于腾格里沙漠土壤(<3.0%)[17].与其他研究类似[15,17],子囊菌门真菌是荒漠土壤中的优势真菌,本研究也发现一些其他未分类真菌门在天然荒漠中也占有较高的相对丰度(图2).本研究中荒漠土壤广古菌门古菌的相对丰度(97.0%)远高于北非的Tataouine 沙漠土壤(40.0%)[45].在Atacama 荒漠、Negev沙漠和内蒙古荒漠草原,土壤真菌群落以曲霉菌属(Aspergillus)和青霉菌属(Penicillium)为主[49-51],而本研究中未分类的真菌属在荒漠土壤真菌群落中占绝对优势(图5C).此外,荒漠土壤中还发现了一些未分类的微生物属(图5),有关这些特殊微生物分类方面的研究还需要进一步加强.可见,盐渍化、极干旱的荒漠环境孕育了有别于其他干旱地区的土壤微生物群落结构. ...
Toward an ecological classification of soil bacteria
1
2007
... 本研究中的农田开垦时间仅仅只有短短的5年,但开垦仍然导致土壤微生物群落结构的明显变化,反映出荒漠土壤微生物群落结构对土地利用方式变化的快速响应[8].整体而言,土壤微生物群落结构与植被属性(盖度、生物量和多样性)、土壤EC、TN、TOC、TP和AP显著相关(表3),其强弱关系因微生物类群的不同而异(图5),可能主要由微生物群落的生物学特性所决定.微生物营养假设认为,农业耕作中由于肥料的使用、根系生物量和分泌物等底物的增加,可能会促使一些富营养型微生物相对丰度的增加,而一些贫营养型微生物的丰度降低[30,52].但是,本研究的结果并不支持这一假设.例如,变形菌门和放线菌门往往被认为是富营养型细菌,而酸杆菌门和硝化螺旋菌门(Nitrospirae)则为贫营养型细菌.而此研究中放线菌门、酸杆菌门和硝化螺旋菌门细菌的相对丰度在农田土壤中都显著提高(图3A),这一结果也表明还有其他因素比养分水平在更大程度上决定着因土地利用方式而导致的土壤微生物群落结构的变化,有关这方面的机理还需要进一步的深入研究.同时,最新的一项研究发现,盐分是荒漠土壤细菌和古菌群落结构的关键影响因子,一些耐盐性的细菌和古菌群落存在于盐渍化的荒漠土壤中[14].因此,荒漠在开垦为农田后由于土壤EC的显著减弱,一些耐盐型的微生物,如γ变形菌门(Gammaproteobacteria)细菌和盐杆菌门(Halobacteria)古菌的相对丰度也随之降低.此外,区域尺度[15,53]和控制试验[54]方面的研究发现,水分是影响荒漠土壤微生物群落结构的关键因子.然而,本研究中荒漠和农田土壤的水分没有显著性差异,Mantel test结果也发现土壤水分与微生物群落结构并不相关,表明该区域短期的开垦过程中土壤水分对土壤微生物群落结构的直接作用并不明显,主要是通过影响植被盖度、生物量和土壤EC间接影响土壤微生物的群落构建[15,34].尽管Mantel test分析结果显示土壤TOC和TN仅与细菌群落结构显著正相关,而与古菌和真菌群落结构并不相关(表3),棉田和荒漠土壤之间的TOC和TN也无显著差异(表1),但长期耕作后势必会导致土壤TOC和TN的进一步显著增加[8].众多研究[17,21-23,27]表明长期的施肥和耕作显著改变了土壤微生物群落结构,由此可以预见荒漠农田土壤微生物群落结构可能也势必会进一步发生变化,但由于缺少连续多年的土壤水分、施肥制度和土壤微生物群落变化的相关数据,还无法建立土壤水分动态、施肥制度与土壤微生物群落结构之间的关系.因此,急需通过开展长时间序列的连续动态监测试验进行荒漠土壤微生物群落演替过程及其作用机理方面的研究. ...
Assessment of the spatial distribution of soil microbial communities in patchy arid and semi-arid landscapes of the Negev Desert using combined PLFA and DGGE analyses
1
2011
... 本研究中的农田开垦时间仅仅只有短短的5年,但开垦仍然导致土壤微生物群落结构的明显变化,反映出荒漠土壤微生物群落结构对土地利用方式变化的快速响应[8].整体而言,土壤微生物群落结构与植被属性(盖度、生物量和多样性)、土壤EC、TN、TOC、TP和AP显著相关(表3),其强弱关系因微生物类群的不同而异(图5),可能主要由微生物群落的生物学特性所决定.微生物营养假设认为,农业耕作中由于肥料的使用、根系生物量和分泌物等底物的增加,可能会促使一些富营养型微生物相对丰度的增加,而一些贫营养型微生物的丰度降低[30,52].但是,本研究的结果并不支持这一假设.例如,变形菌门和放线菌门往往被认为是富营养型细菌,而酸杆菌门和硝化螺旋菌门(Nitrospirae)则为贫营养型细菌.而此研究中放线菌门、酸杆菌门和硝化螺旋菌门细菌的相对丰度在农田土壤中都显著提高(图3A),这一结果也表明还有其他因素比养分水平在更大程度上决定着因土地利用方式而导致的土壤微生物群落结构的变化,有关这方面的机理还需要进一步的深入研究.同时,最新的一项研究发现,盐分是荒漠土壤细菌和古菌群落结构的关键影响因子,一些耐盐性的细菌和古菌群落存在于盐渍化的荒漠土壤中[14].因此,荒漠在开垦为农田后由于土壤EC的显著减弱,一些耐盐型的微生物,如γ变形菌门(Gammaproteobacteria)细菌和盐杆菌门(Halobacteria)古菌的相对丰度也随之降低.此外,区域尺度[15,53]和控制试验[54]方面的研究发现,水分是影响荒漠土壤微生物群落结构的关键因子.然而,本研究中荒漠和农田土壤的水分没有显著性差异,Mantel test结果也发现土壤水分与微生物群落结构并不相关,表明该区域短期的开垦过程中土壤水分对土壤微生物群落结构的直接作用并不明显,主要是通过影响植被盖度、生物量和土壤EC间接影响土壤微生物的群落构建[15,34].尽管Mantel test分析结果显示土壤TOC和TN仅与细菌群落结构显著正相关,而与古菌和真菌群落结构并不相关(表3),棉田和荒漠土壤之间的TOC和TN也无显著差异(表1),但长期耕作后势必会导致土壤TOC和TN的进一步显著增加[8].众多研究[17,21-23,27]表明长期的施肥和耕作显著改变了土壤微生物群落结构,由此可以预见荒漠农田土壤微生物群落结构可能也势必会进一步发生变化,但由于缺少连续多年的土壤水分、施肥制度和土壤微生物群落变化的相关数据,还无法建立土壤水分动态、施肥制度与土壤微生物群落结构之间的关系.因此,急需通过开展长时间序列的连续动态监测试验进行荒漠土壤微生物群落演替过程及其作用机理方面的研究. ...
Water regime history drives responses of soil Namib Desert microbial communities to wetting events
1
2015
... 本研究中的农田开垦时间仅仅只有短短的5年,但开垦仍然导致土壤微生物群落结构的明显变化,反映出荒漠土壤微生物群落结构对土地利用方式变化的快速响应[8].整体而言,土壤微生物群落结构与植被属性(盖度、生物量和多样性)、土壤EC、TN、TOC、TP和AP显著相关(表3),其强弱关系因微生物类群的不同而异(图5),可能主要由微生物群落的生物学特性所决定.微生物营养假设认为,农业耕作中由于肥料的使用、根系生物量和分泌物等底物的增加,可能会促使一些富营养型微生物相对丰度的增加,而一些贫营养型微生物的丰度降低[30,52].但是,本研究的结果并不支持这一假设.例如,变形菌门和放线菌门往往被认为是富营养型细菌,而酸杆菌门和硝化螺旋菌门(Nitrospirae)则为贫营养型细菌.而此研究中放线菌门、酸杆菌门和硝化螺旋菌门细菌的相对丰度在农田土壤中都显著提高(图3A),这一结果也表明还有其他因素比养分水平在更大程度上决定着因土地利用方式而导致的土壤微生物群落结构的变化,有关这方面的机理还需要进一步的深入研究.同时,最新的一项研究发现,盐分是荒漠土壤细菌和古菌群落结构的关键影响因子,一些耐盐性的细菌和古菌群落存在于盐渍化的荒漠土壤中[14].因此,荒漠在开垦为农田后由于土壤EC的显著减弱,一些耐盐型的微生物,如γ变形菌门(Gammaproteobacteria)细菌和盐杆菌门(Halobacteria)古菌的相对丰度也随之降低.此外,区域尺度[15,53]和控制试验[54]方面的研究发现,水分是影响荒漠土壤微生物群落结构的关键因子.然而,本研究中荒漠和农田土壤的水分没有显著性差异,Mantel test结果也发现土壤水分与微生物群落结构并不相关,表明该区域短期的开垦过程中土壤水分对土壤微生物群落结构的直接作用并不明显,主要是通过影响植被盖度、生物量和土壤EC间接影响土壤微生物的群落构建[15,34].尽管Mantel test分析结果显示土壤TOC和TN仅与细菌群落结构显著正相关,而与古菌和真菌群落结构并不相关(表3),棉田和荒漠土壤之间的TOC和TN也无显著差异(表1),但长期耕作后势必会导致土壤TOC和TN的进一步显著增加[8].众多研究[17,21-23,27]表明长期的施肥和耕作显著改变了土壤微生物群落结构,由此可以预见荒漠农田土壤微生物群落结构可能也势必会进一步发生变化,但由于缺少连续多年的土壤水分、施肥制度和土壤微生物群落变化的相关数据,还无法建立土壤水分动态、施肥制度与土壤微生物群落结构之间的关系.因此,急需通过开展长时间序列的连续动态监测试验进行荒漠土壤微生物群落演替过程及其作用机理方面的研究. ...





 甘公网安备 62010202000688号
甘公网安备 62010202000688号
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}